 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ems | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE C. ELEGANS NITFHIT PROTEIN | ||||||
 Components Components | NIT-FRAGILE HISTIDINE TRIAD FUSION PROTEIN | ||||||
 Keywords Keywords | ANTITUMOR PROTEIN / WORM / NITRILASE / FHIT / NUCLEOTIDE-BINDING PROTEIN / CANCER / DIADENOSINE POLYPHOSPHATE HYDROLASE / HISTIDINE TRIAD / TUMOR SUPPRESSOR / ROSETTA STONE | ||||||
| Function / homology |  Function and homology information Function and homology informationdeaminated glutathione amidase activity / bis(5'-adenosyl)-triphosphatase / bis(5'-adenosyl)-triphosphatase activity / Hydrolases; Acting on carbon-nitrogen bonds, other than peptide bonds / : / nucleobase-containing compound metabolic process / nucleotide binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / TWO WAVELENGTH ANOMALOUS DIFFRACTION / Resolution: 2.8 Å X-RAY DIFFRACTION / SYNCHROTRON / TWO WAVELENGTH ANOMALOUS DIFFRACTION / Resolution: 2.8 Å | ||||||
 Authors Authors | Pace, H.C. / Hodawadekar, S.C. / Draganescu, A. / Huang, J. / Bieganowski, P. / Pekarsky, Y. / Croce, C.M. / Brenner, C. | ||||||
 Citation Citation | Journal: Curr.Biol. / Year: 2000 Title: Crystal structure of the worm NitFhit Rosetta Stone protein reveals a Nit tetramer binding two Fhit dimers. Authors: Pace, H.C. / Hodawadekar, S.C. / Draganescu, A. / Huang, J. / Bieganowski, P. / Pekarsky, Y. / Croce, C.M. / Brenner, C. #1: Journal: Proc.Natl.Acad.Sci.USA / Year: 1998Title: Nitrilase and Fhit homologs are encoded as fusion proteins in Drosophila melanogaster and Caenorhabditis elegans Authors: Pekarsky, Y. / Campiglio, M. / Siprashvili, Z. / Druck, T. / Sedkov, Y. / Tillib, S. / Draganescu, A. / Wermuth, P. / Rothman, J.H. / Huebner, K. / Buchberg, A.M. / Mazo, A. / Brenner, C. / Croce, C.M. #2: Journal: Proc.Natl.Acad.Sci.USA / Year: 1998Title: Genetic, biochemical and crystallographic characterization of Fhit-substrate complexes as the active signaling form of Fhit Authors: Pace, H.C. / Garrison, P.N. / Robinson, A.K. / Barnes, L.D. / Draganescu, A. / Rosler, A. / Blackburn, G.M. / Siprashvili, Z. / Croce, C.M. / Huebner, K. / Brenner, C. #3: Journal: J.CELL PHYSIOL. / Year: 1999Title: The Histidine Triad Superfamily of Nucleotide-Binding Proteins Authors: Brenner, C. / Bieganowski, P. / Pace, H.C. / Huebner, K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ems.cif.gz | 174.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ems.ent.gz | 139.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1ems.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/em/1emsftp://data.pdbj.org/pub/pdb/validation_reports/em/1ems | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|






-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a tetramer constructed from the crystallographic symmetry partners of chains A and B |
-Components
| #1: Protein | Mass: 50007.945 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Chemical | ChemComp-EMC / 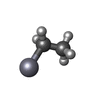  Mass: 229.651 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C2H5Hg Mass: 229.651 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C2H5Hg#3: Chemical | ChemComp-NA /   Mass: 22.990 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Na#4: Chemical | ChemComp-MPD / ( |   Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 161 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 161 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.74 Å3/Da / Density % sol: 55.06 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 7 Details: methylpentanediol, sodium chloride, HEPES, pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 295.0K | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 98 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X8C / Wavelength: 1.0090, 0.9928 / Beamline: X8C / Wavelength: 1.0090, 0.9928 | |||||||||
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Jul 11, 1999 | |||||||||
| Radiation | Protocol: TWO WAVELENGTH ANOMALOUS DIFFRACTION / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 2.8→40 Å / Num. all: 49928 / Num. obs: 49928 / % possible obs: 95.2 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 7.5 % / Rmerge(I) obs: 0.045 / Net I/σ(I): 24.6 | |||||||||
| Reflection shell | Resolution: 2.8→2.9 Å / Rmerge(I) obs: 0.088 / Num. unique all: 3897 | |||||||||
| Reflection | *PLUS Lowest resolution: 30 Å / % possible obs: 97.9 % | |||||||||
| Reflection shell | *PLUS % possible obs: 91.4 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: TWO WAVELENGTH ANOMALOUS DIFFRACTION Resolution: 2.8→30 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 5410833.97 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber / Details: mlhl target and bulk solvent model used
| |||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 27.7963 Å2 / ksol: 0.343798 e/Å3 | |||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28.5 Å2
| |||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→30 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.8→2.9 Å / Rfactor Rfree error: 0.019 / Total num. of bins used: 10
| |||||||||||||||||||||||||
| Xplor file |
| |||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.9 / Classification: refinement | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
