Mass: 18.015 Da / Num. of mol.: 266 / Source method: isolated from a natural source / Formula: H2O
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 3
-
Sample preparation
Crystal
Density Matthews: 3.79 Å3/Da / Density % sol: 62 % Description: STRUCTURE DETERMINED USING THE SULFUR SAS SIGNAL RECORDED AT ID17, APS USING 1.74 ANGSTROM X-RAYS (SEE REMARK 3 REFINEMENT DETAILS)
Crystal grow
Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 6 Details: HANGING DROP VAPOR DIFFUSION USING 5 MICROLITER DROPS CONTAINING EQUAL VOLUMES OF PROTEIN CONCENTRATE (10 MG/ML) AND RESERVOIR SOLUTION: CONTAINING 1.4 M SODIUM CITRATE, PH 6.0., VAPOR ...Details: HANGING DROP VAPOR DIFFUSION USING 5 MICROLITER DROPS CONTAINING EQUAL VOLUMES OF PROTEIN CONCENTRATE (10 MG/ML) AND RESERVOIR SOLUTION: CONTAINING 1.4 M SODIUM CITRATE, PH 6.0., VAPOR DIFFUSION, HANGING DROP, temperature 277K
Method to determine structure: SULFUR SAS / Resolution: 1.73→19.84 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 260910.23 / Data cutoff low absF: 0 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber Details: THE STRUCTURE WAS DETERMINED USING SINGLE WAVELENGTH ANOMALOUS SCATTERING DATA RECORDED FOR THE NATIVE PROTEIN. DATA WERE RECORDED ON TWO OBELIN CRYSTALS TO 2.5 ANGSTROMS RESOLUTION AT ...Details: THE STRUCTURE WAS DETERMINED USING SINGLE WAVELENGTH ANOMALOUS SCATTERING DATA RECORDED FOR THE NATIVE PROTEIN. DATA WERE RECORDED ON TWO OBELIN CRYSTALS TO 2.5 ANGSTROMS RESOLUTION AT BEAMLINE ID-17 (IMCA-CAT), ADVANCED PHOTON SOURCE (APS), ARGONNE NATIONAL LABORATORY USING A MARRESEARCH 165 MM CCD DETECTOR AND 1.74 ANGSTROM X-RAYS. A DATA COLLECTION STRATEGY (HKL2000) WAS DETERMINED FROM THE INITIAL IMAGE TO GIVE THE STARTING AND ENDING POINTS OF THE DATA COLLECTION TO INSURE A MINIMUM DATA COMPLETENESS OF 98 PERCENT. DATA WERE COLLECTED USING THE "FREIDEL FLIP" METHOD. DATA PROCESSING WAS CARRIED OUT USING HKL2000. THE PROCESSED DATA FORMING THE TWO DATA SETS WERE MERGED TO INCREASE REDUNDANCY (WU, ET AL., (2000) J. PROT. PEP. LETTS. 7, 25-32) AND SOLVE WAS USED TO DETERMINE THE SIX OF THE EIGHT SULFUR ATOM POSITIONS. PHASES WAS USED TO REFINE THE SULFUR POSITIONS AND TO LOCATE THE REMAINING SULFUR SITES BY BIJVOET DIFFERENCE FOURIER ANALYSIS. THREE ADDITIONAL SITES WERE IDENTIFIED SUGGESTING THAT ONE OF THE SULFUR SITES WAS MOST PROBABLY AN ANOMALOUS SOLVENT SUCH AS CHLORIDE. ALL ANOMALOUS SITES WERE TREATED AS SULFUR FOR THE PHASE CALCULATIONS. THE NINE SULFUR SITES WERE USED TO ESTIMATE THE INITIAL PROTEIN PHASES AT 3 ANGSTROM RESOLUTION USING SOLVENT FLATTENING (ISAS, WANG, (1985) METHODS ENZYMOL. 115, 90 112). A PRELIMINARY CHAIN TRACE WAS DONE USING THE 3.0 ANGSTROM MAP. SEQUENCE ASSIGNMENT WAS BASED ON 8 SULFUR ATOM POSITIONS. THE MODEL, 99 PERCENT COMPLETE WAS BUILT USING THE 3.0 ANGSTROM SAS MAP. RESIDUE 1 WAS NOT OBSERVED IN THE ELECTRON DENSITY AMPS AND IS PRESUMED TO BE DISORDERED. THE RESOLUTION OF THE STRUCTURE WAS THEN EXTENDED TO 1.73 ANGSTROMS RESOLUTION DURING THE FINAL STAGES OF REFINEMENT USING A MERGED DATA SET COLLECTED FROM A SINGLE CRYSTAL RECORDED AT APS (BEAMLINE ID19, WAVELENGTH 0.94 ANGSTROMS) AND AT ALS (BEAMLINE 5.0.2, WAVELENGTH 1.0 ANGSTROMS).
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
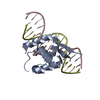
Function and homology information Obelia longissima (invertebrata)
Obelia longissima (invertebrata) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








 PDBj
PDBj Assembly
Assembly


 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl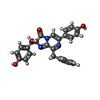
 Mass: 439.463 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C26H21N3O4
Mass: 439.463 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C26H21N3O4 Mass: 18.015 Da / Num. of mol.: 266 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 266 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation
 Processing
Processing