 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1e0b | ||||||
|---|---|---|---|---|---|---|---|
| Title | Chromo shadow domain from fission yeast swi6 protein. | ||||||
 Components Components | SWI6 PROTEIN | ||||||
 Keywords Keywords | CHROMATIN-BINDING / CHROMODOMAIN / SHADOW / HETEROCHROMATIN / SWI6 / POMBE | ||||||
| Function / homology |  Function and homology information Function and homology informationsiRNA-mediated silent mating type cassette region heterochromatin formation / meiotic centromeric cohesion protection in anaphase I / co-transcriptional gene silencing by RNA interference machinery / gene conversion at mating-type locus / mitotic telomere tethering at nuclear periphery / donor selection / mating type switching / heterochromatin island / subtelomeric heterochromatin / mitotic sister chromatid cohesion, centromeric ...siRNA-mediated silent mating type cassette region heterochromatin formation / meiotic centromeric cohesion protection in anaphase I / co-transcriptional gene silencing by RNA interference machinery / gene conversion at mating-type locus / mitotic telomere tethering at nuclear periphery / donor selection / mating type switching / heterochromatin island / subtelomeric heterochromatin / mitotic sister chromatid cohesion, centromeric / heterochromatin boundary formation / mitotic sister chromatid biorientation / mating-type region heterochromatin / pericentric heterochromatin formation / siRNA-mediated pericentric heterochromatin formation / condensed chromosome, centromeric region / silent mating-type cassette heterochromatin formation / chromatin-protein adaptor activity / histone H3K9me2/3 reader activity / subtelomeric heterochromatin formation / pericentric heterochromatin / heterochromatin / chromatin organization / heterochromatin formation / chromatin binding / chromatin / DNA-templated transcription / DNA binding / RNA binding / identical protein binding / nucleus Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MIR / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MIR / Resolution: 1.9 Å | ||||||
 Authors Authors | Cowieson, N.P. / Partridge, J.F. / Allshire, R.C. / Mclaughlin, P.J. | ||||||
 Citation Citation | Journal: Curr.Biol. / Year: 2000 Title: Dimerisation of Chromo Shadow Domain and Distinctions from the Chromodomain as Revealed by Structural Analysis Authors: Cowieson, N.P. / Partridge, J.F. / Allshire, R.C. / Mclaughlin, P.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1e0b.cif.gz | 39.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1e0b.ent.gz | 28.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1e0b.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/e0/1e0bftp://data.pdbj.org/pub/pdb/validation_reports/e0/1e0b | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.91, -0.408, -0.079), Vector: |
-Components
| #1: Protein | Mass: 8070.922 Da / Num. of mol.: 2 / Fragment: CHROMO SHADOW DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Description: RECOMBINANT PROTEIN OVEREXPRESSED IN ESCHERICHIA COLI. Production host:  #2: Chemical | ChemComp-1PG / | 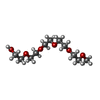  Mass: 252.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H24O6 Mass: 252.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H24O6#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 111 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 111 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.93 Å3/Da / Density % sol: 61 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / pH: 8.5 Details: 20% (W/V) PEG 4000, 0.2M SODIUM ACETATE, 0.1M TRIS HCL PH 8.5, TEMPERATURE 4 DEGREES CENTIGRADE PROTEIN CONCENTRATION 10 MG/ML TIME 3 TO 4 DAYS | |||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.6 / Wavelength: 0.87 / Beamline: PX9.6 / Wavelength: 0.87 |
| Detector | Date: May 15, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.87 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→22.25 Å / Num. obs: 115148 / % possible obs: 99.8 % / Redundancy: 14 % / Rmerge(I) obs: 0.061 |
| Reflection | *PLUS Num. measured all: 115148 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIR / Resolution: 1.9→22 Å / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 41.6546 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→22 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.9 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 41.6546 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
