 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1do1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CARBONMONOXY-MYOGLOBIN MUTANT L29W AT 105K | |||||||||
 Components Components | MYOGLOBIN | |||||||||
 Keywords Keywords | OXYGEN STORAGE/TRANSPORT / HEME / RESPIRATORY PROTEIN / OXYGEN STORAGE-TRANSPORT COMPLEX | |||||||||
| Function / homology |  Function and homology information Function and homology informationOxidoreductases; Acting on other nitrogenous compounds as donors / nitrite reductase activity / sarcoplasm / Oxidoreductases; Acting on a peroxide as acceptor; Peroxidases / removal of superoxide radicals / oxygen carrier activity / peroxidase activity / oxygen binding / heme binding / extracellular exosome / metal ion binding Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.5 Å X-RAY DIFFRACTION / Resolution: 1.5 Å | |||||||||
 Authors Authors | Ostermann, A. / Waschipky, R. / Parak, F.G. / Nienhaus, G.U. | |||||||||
 Citation Citation | Journal: Nature / Year: 2000 Title: Ligand binding and conformational motions in myoglobin. Authors: Ostermann, A. / Waschipky, R. / Parak, F.G. / Nienhaus, G.U. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1do1.cif.gz | 52 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1do1.ent.gz | 35 KB | Display | PDB format |
| PDBx/mmJSON format | 1do1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/do/1do1ftp://data.pdbj.org/pub/pdb/validation_reports/do/1do1 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1do3C  1do4C  1do7C  2mbwS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
-Protein / Sugars , 2 types, 2 molecules A
| #1: Protein | Mass: 17466.225 Da / Num. of mol.: 1 / Mutation: M0(FME), L29W, D122N Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|---|
| #2: Polysaccharide | alpha-D-glucopyranose-(1-1)-alpha-D-glucopyranose / trehalose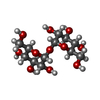  Source method: isolated from a genetically manipulated source Details: oligosaccharide with reducing-end-to-reducing-end glycosidic bond References: trehalose |
-Non-polymers , 4 types, 195 molecules 

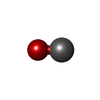

| #3: Chemical |  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-HEM / |  Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4#5: Chemical | ChemComp-CMO / |  Mass: 28.010 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CO Mass: 28.010 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CO#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 191 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | N |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.06 Å3/Da / Density % sol: 59.8 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 292 K / pH: 8.5 Details: CRYSTALS WERE GROWN IN 2.5M AMMONIUM SULFATE SOLUTION, BUFFERED WITH 20MM TRIS/ HCL TO PH 8.5. THE CRYSTALLIZATION SOLUTION WAS REPLACED IN STEPS AGAINST A SOLUTION CONTAINING 2.5M AMMONIUM ...Details: CRYSTALS WERE GROWN IN 2.5M AMMONIUM SULFATE SOLUTION, BUFFERED WITH 20MM TRIS/ HCL TO PH 8.5. THE CRYSTALLIZATION SOLUTION WAS REPLACED IN STEPS AGAINST A SOLUTION CONTAINING 2.5M AMMONIUM SULFATE, 20MM TRIS, 300MG/ML TREHALOSE AT PH 8.5., temperature 292K | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7.5 / Method: unknown | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 105 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ENRAF-NONIUS FR591 / Wavelength: 1.54 |
| Detector | Type: SIEMENS HI-STAR / Detector: AREA DETECTOR / Date: Nov 3, 1998 / Details: graphite |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→10 Å / Num. obs: 32053 / % possible obs: 94.5 % / Observed criterion σ(I): 2 / Redundancy: 5.5 % / Biso Wilson estimate: 9.7 Å2 / Rmerge(I) obs: 0.043 / Net I/σ(I): 14.6 |
| Reflection shell | Resolution: 1.5→1.55 Å / Redundancy: 3.1 % / Rmerge(I) obs: 0.139 / Mean I/σ(I) obs: 5.1 / % possible all: 82.3 |
| Reflection | *PLUS Num. measured all: 175770 |
| Reflection shell | *PLUS % possible obs: 82.3 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Starting model: 2MBW Resolution: 1.5→7 Å / Data cutoff high absF: 10000000 / Data cutoff low absF: 0 / Cross valid method: free r / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: NO ANGLE RESTRAINTS WERE USED FOR THE CO MOLECULE AND THE HIS 93 WITH RESPECT TO THE IRON ATOM. BOND RESTRAINTS FOR THE CO MOLECULE, HIS 93 AND FOR THE PYRROLE NITROGEN TO THE IRON ATOM WERE ...Details: NO ANGLE RESTRAINTS WERE USED FOR THE CO MOLECULE AND THE HIS 93 WITH RESPECT TO THE IRON ATOM. BOND RESTRAINTS FOR THE CO MOLECULE, HIS 93 AND FOR THE PYRROLE NITROGEN TO THE IRON ATOM WERE WEAKENED FROM THE STANDARD X-PLOR VALUES (PARAM19X.HEME). THERE IS NEARLY NO ELECTRON DENSITY FOR THE N-FORMYL-MET. THIS RESIDUE WAS NOT INCLUDED INTO THE MODEL. ELECTRON DENSITY FEATURES CLOSE TO THIS SITE WERE MODELED BY WATER MOLECULES. ONE TREHALOSE MOLECULE IS BOUND TO THE SURFACE OF THE MYOGLOBIN MOLECULE. THE WATER MOLECULES NUMBER 246 AND 252 ARE ON OR CLOSE TO SPECIAL POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati d res low obs: 7 Å / Luzzati sigma a obs: 0.14 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.5→1.57 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.5 Å / Lowest resolution: 7 Å / σ(F): 0 / Rfactor obs: 0.189 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.257 / Rfactor Rwork: 0.236 |
