 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cw9 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | DNA DECAMER WITH AN ENGINEERED CROSSLINK IN THE MINOR GROOVE | ||||||||||||||||||
 Components Components | 5'-D(* Keywords KeywordsDNA / CROSSLINKED DOUBLE-HELICAL DNA / BASE-PAIR OPENING / PARTIAL BASE FLIPPING. | Function / homology | DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.55 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.55 Å  Authors Authorsvan Aalten, D.M.F. / Erlanson, D.A. / Verdine, G.L. / Joshua-Tor, L. |  CitationJournal: Proc.Natl.Acad.Sci.USA / Year: 1999 CitationJournal: Proc.Natl.Acad.Sci.USA / Year: 1999Title: A structural snapshot of base-pair opening in DNA. Authors: van Aalten, D.M. / Erlanson, D.A. / Verdine, G.L. / Joshua-Tor, L. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cw9.cif.gz | 38.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cw9.ent.gz | 26.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1cw9.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1cw9_validation.pdf.gz | 341.2 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1cw9_full_validation.pdf.gz | 341.7 KB | Display | |
| Data in XML | 1cw9_validation.xml.gz | 2.1 KB | Display | |
| Data in CIF | 1cw9_validation.cif.gz | 4.4 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cw/1cw9ftp://data.pdbj.org/pub/pdb/validation_reports/cw/1cw9 | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|





-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 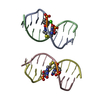
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 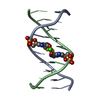
| ||||||||||
| 2 | 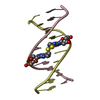
| ||||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 3106.110 Da / Num. of mol.: 4 / Source method: obtained synthetically Details: CROSSLINKED DNA WAS SYNTHESIZED BY THE CONVERTIBLE NUCLEOSIDE APPROACH #2: Chemical | ChemComp-CA / |   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 271 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 271 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.13 Å3/Da / Density % sol: 45 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 7 Details: MPD, CALCIUM CHLORIDE, SODIUM CHLORIDE, SPERMINE TETRACHLORIDE, SODIUM CACODYLATE, pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 277K | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X26C / Wavelength: 1.08 / Beamline: X26C / Wavelength: 1.08 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Mar 2, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.08 Å / Relative weight: 1 |
| Reflection | Resolution: 1.55→13 Å / Num. all: 62990 / Num. obs: 62990 / % possible obs: 95.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4 % / Biso Wilson estimate: 14.7 Å2 / Rmerge(I) obs: 0.078 / Net I/σ(I): 9 |
| Reflection shell | Resolution: 1.55→1.61 Å / Redundancy: 4 % / Rmerge(I) obs: 0.32 / Mean I/σ(I) obs: 3 / % possible all: 98 |
| Reflection | *PLUS Num. obs: 15570 / Num. measured all: 62990 |
| Reflection shell | *PLUS % possible obs: 98 % / Num. unique obs: 1555 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: BDJ017 Resolution: 1.55→13 Å / Isotropic thermal model: RESTRAINED CNS / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 Stereochemistry target values: CNS NUCLEIC ACID PARAMETERS USING SOFT VAN DER WAALS PARAMETERS (PARKINSON ET AL., ACTA D, 52, 57-64 (1996)). Details: MAXIMUM LIKELIHOOD.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT - CNS CALCULATED MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 18.3 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.55→13 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.55→1.61 Å / % reflection Rfree: 5 % / % reflection obs: 98 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file | Serial no: 1 / Param file: DNA-RNA_REP.PARAM / Topol file: DNA-RNA.TOP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 13 Å / σ(F): 0 / % reflection Rfree: 5 % / Rfactor obs: 0.195 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 18.3 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS % reflection Rfree: 5 % |
