 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ctf: STRUCTURE OF THE C-TERMINAL DOMAIN OF THE RIBOSOMAL PROTEIN L7/L1... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ctf | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF THE C-TERMINAL DOMAIN OF THE RIBOSOMAL PROTEIN L7/L12 FROM ESCHERICHIA COLI AT 1.7 ANGSTROMS | ||||||
 Components Components | RIBOSOMAL PROTEIN L7/L12 | ||||||
 Keywords Keywords | RIBOSOMAL PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationlarge ribosomal subunit / ribosome binding / cytosolic large ribosomal subunit / cytoplasmic translation / structural constituent of ribosome / translation / mRNA binding / protein homodimerization activity / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.7 Å X-RAY DIFFRACTION / Resolution: 1.7 Å | ||||||
 Authors Authors | Leijonmarck, M. / Liljas, A. | ||||||
 Citation Citation | Journal: J Mol Biol / Year: 1987 Title: Structure of the C-terminal domain of the ribosomal protein L7/L12 from Escherichia coli at 1.7 A. Authors: M Leijonmarck / A Liljas /  Abstract: The structure of a C-terminal fragment of the ribosomal protein L7/L12 from Escherichia coli has been refined using crystallographic data to 1.7 A resolution. The R-value is 17.4%. Six residues at ...The structure of a C-terminal fragment of the ribosomal protein L7/L12 from Escherichia coli has been refined using crystallographic data to 1.7 A resolution. The R-value is 17.4%. Six residues at the N terminus are too disordered in the structure to be localized. These residues are probably part of a hinge in the complete L7/L12 molecule. The possibility that a 2-fold crystallographic axis is a molecular 2-fold axis is discussed. A patch of invariant residues on the surface of the dimer is probably involved in functional interactions with elongation factors. #1: Journal: Structural Aspects of Recognition and Assembly in Biological MacromoleculesYear: 1981 Title: Structural Studies on the Protein L7(Slash)L12 from E. Coli Ribosomes Authors: Leijonmarck, M. / Pettersson, I. / Liljas, A. #2: Journal: Nature / Year: 1980Title: Crystal Structure of a Ribosomal Component at 2.6 Angstroms Resolution Authors: Leijonmarck, M. / Eriksson, S. / Liljas, A. #3: Journal: FEBS Lett. / Year: 1978Title: Isolation and Crystallization of Stable Domains of the Protein L7(Slash)L12 from Escherichia Coli Ribosomes Authors: Liljas, A. / Eriksson, S. / Donner, D. / Kurland, C.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ctf.cif.gz | 24.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ctf.ent.gz | 16 KB | Display | PDB format |
| PDBx/mmJSON format | 1ctf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ct/1ctfftp://data.pdbj.org/pub/pdb/validation_reports/ct/1ctf | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 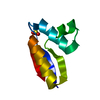
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Atom site foot note | 1: THE ELECTRON DENSITY FOR RESIDUE LYS 59 IS POORLY DEFINED FROM CD TO NZ. 2: THE ELECTRON DENSITY FOR RESIDUES LYS 81, LYS 108, LYS 120 IS POORLY DEFINED FROM CG TO NZ. 3: RESIDUE PRO 91 IS A CIS PROLINE. | |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 7573.641 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 62 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 62 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.12 Å3/Da / Density % sol: 41.9 % |
|---|---|
| Crystal grow | *PLUS Method: other / Details: Leijonmarck, M., (1980) Nature, 286, 824. |
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 1.69 Å / Num. all: 8688 / Num. obs: 7863 / Rmerge(I) obs: 0.018 |
- Processing
Processing
| Software | Name: CORELS / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Highest resolution: 1.7 Å /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 1.7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 6.02 Å / Rfactor all: 0.196 / Rfactor obs: 0.174 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
