 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1cpx: BETA FORM OF CARBOXYPEPTIDASE A (RESIDUES 3-307) FROM BOVINE PANC... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cpx | ||||||
|---|---|---|---|---|---|---|---|
| Title | BETA FORM OF CARBOXYPEPTIDASE A (RESIDUES 3-307) FROM BOVINE PANCREAS IN AN ORTHORHOMBIC CRYSTAL FORM WITH TWO ZINC IONS IN THE ACTIVE SITE. | ||||||
 Components Components | PROTEIN (CARBOXYPEPTIDASE A) | ||||||
 Keywords Keywords | HYDROLASE / METALLOPROTEASE / CARBOXYPEPTIDASE / ZINC INHIBITION / INDUCED FIT | ||||||
| Function / homology |  Function and homology information Function and homology informationcarboxypeptidase A / leukotriene metabolic process / metallocarboxypeptidase activity / proteolysis / : / zinc ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Bukrinsky, J.T. / Bjerrum, M.J. / Kadziola, A. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1998 Title: Native carboxypeptidase A in a new crystal environment reveals a different conformation of the important tyrosine 248. Authors: Bukrinsky, J.T. / Bjerrum, M.J. / Kadziola, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cpx.cif.gz | 79 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cpx.ent.gz | 58.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1cpx.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cp/1cpxftp://data.pdbj.org/pub/pdb/validation_reports/cp/1cpx | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5cpaS S: Starting model for refinement |
|---|---|
| Similar structure data |
















-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34445.418 Da / Num. of mol.: 1 / Source method: isolated from a natural source Details: SIGMA #C0261; COX ET AL. (1964) BIOCHEMISTRY 3, 44-47 Source: (natural) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#3: Chemical | ChemComp-OH / | 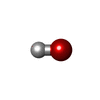  Mass: 17.007 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: HO Mass: 17.007 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: HO#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 277 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 277 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Nonpolymer details | ZN 308 IS THE CATALYTIC ZINC AND ZN 309 IS THE INHIBITORY ZINC OH 541 IS THE HYDROXIDE ION, WHICH ...ZN 308 IS THE CATALYTIC ZINC AND ZN 309 IS THE INHIBITORY | Sequence details | SER 3 - ASN 307, I.E. THE BETA FORM OF CPA | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.25 Å3/Da / Density % sol: 45 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: microdialysis / pH: 7.5 Details: CPA FROM SIGMA #C0261 WAS DISSOLVED IN 1.0 M LICL WITH 20 MM TRIS-HCL PH 7.5 TO GIVE 12 MG/ML AND USED WITHOUT FURTHER PURIFICATION. THIS PROTEIN SOLUTION WAS PLACED IN 50 MICROL DIALYSIS ...Details: CPA FROM SIGMA #C0261 WAS DISSOLVED IN 1.0 M LICL WITH 20 MM TRIS-HCL PH 7.5 TO GIVE 12 MG/ML AND USED WITHOUT FURTHER PURIFICATION. THIS PROTEIN SOLUTION WAS PLACED IN 50 MICROL DIALYSIS BUTTONS FROM HAMPTON RESEARCH AND DIALYZED AGAINST A 2 ML RESERVOIR OF THE ABOVE MENTIONED BUFFER WHILE LOWERING THE EXTERNAL LICL CONCENTRATION BY 25% EVERY OTHER DAY. THE CRYSTALS APPEARED A~0.24 M LICL AND WAS STORED AT 0.30M LICL. IF THIS PROCEDURE DOES NOT WORK BY ITSELF AND YOU END UP WITH THE MONOCLINIC ALPHA-FORM (1-307), YOU CAN TRY TO AD TRACES OF TRYPSIN (~10 MICROG/ML) TO THE PROTEIN SOLUTION TO PROVOKE FORMATION OF THE BETA-FORM (3-307), micro dialysis | ||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||
| Crystal grow | *PLUS Method: microdialysis | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 290 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.54 |
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE / Date: Dec 6, 1996 |
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 2→40 Å / Num. obs: 20453 / % possible obs: 94.1 % / Redundancy: 4.3 % / Biso Wilson estimate: 19 Å2 / Rmerge(I) obs: 0.038 / Rsym value: 0.038 / Net I/σ(I): 30.7 |
| Reflection shell | Resolution: 2→2.03 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.157 / Mean I/σ(I) obs: 7.1 / Rsym value: 0.157 / % possible all: 67.7 |
| Reflection shell | *PLUS % possible obs: 67.7 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5CPA WITHOUT WATERS AND ZN Resolution: 2→8 Å / Data cutoff low absF: 0 / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.8 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.09 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 5 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 15.8 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.252 / % reflection Rfree: 4.2 % / Rfactor Rwork: 0.224 |
