 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1cmj: CRYSTAL STRUCTURES OF FERRIC-NO COMPLEXES OF FUNGAL NITRIC OXIDE ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cmj | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURES OF FERRIC-NO COMPLEXES OF FUNGAL NITRIC OXIDE REDUCTASE AND THEIR SER286 MUTANTS AT CRYOGENIC TEMPERATURE | ||||||
 Components Components | CYTOCHROME P450 | ||||||
 Keywords Keywords | OXIDOREDUCTASE / NITRIC OXIDE REDUCTASE / CYTOCHROME P450NOR / NO-LIGANDED / MUTAGENESIS(S286T) | ||||||
| Function / homology |  Function and homology information Function and homology informationnitric oxide reductase [NAD(P)+, nitrous oxide-forming] / nitric oxide reductase [NAD(P)H] activity / oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen / monooxygenase activity / iron ion binding / heme binding Similarity search - Function | ||||||
| Biological species |   Fusarium oxysporum (fungus) Fusarium oxysporum (fungus) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Park, S.-Y. / Shiro, Y. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2000 Title: Proton delivery in NO reduction by fungal nitric-oxide reductase. Cryogenic crystallography, spectroscopy, and kinetics of ferric-NO complexes of wild-type and mutant enzymes. Authors: Shimizu, H. / Obayashi, E. / Gomi, Y. / Arakawa, H. / Park, S.Y. / Nakamura, H. / Adachi, S. / Shoun, H. / Shiro, Y. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cmj.cif.gz | 99.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cmj.ent.gz | 73.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1cmj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cm/1cmjftp://data.pdbj.org/pub/pdb/validation_reports/cm/1cmj | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1cl6C  1cmnC  1romS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |


















-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 44303.520 Da / Num. of mol.: 1 / Mutation: S286T Source method: isolated from a genetically manipulated source Source: (gene. exp.) Fusarium oxysporum (fungus) / Plasmid: PNORST / Gene (production host): CYP55 / Production host:  References: UniProt: P23295, Oxidoreductases; Acting on paired donors, with incorporation or reduction of molecular oxygen |
|---|---|
| #2: Chemical | ChemComp-HEM /   Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 |
| #3: Chemical | ChemComp-NO / 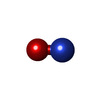  Mass: 30.006 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: NO Mass: 30.006 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: NO |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 337 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 337 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 41 % Description: ROTATION AND TRANSLATION SEARCH IN THE X-PLOR PACKAGE | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7 / Details: 100MM-MES BUFFER AT PH 7.0 USING PEG4K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 5.6 / Method: vapor diffusion, sitting drop / Details: Park, S.Y., (1997) FEBS Lett. 412, 346. | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8  / Beamline: BL44B2 / Wavelength: 0.7 / Beamline: BL44B2 / Wavelength: 0.7 |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Aug 1, 1998 / Details: MIRROR/MONOCHROMATOR |
| Radiation | Monochromator: SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.7 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→100 Å / Num. obs: 42130 / % possible obs: 97.8 % / Observed criterion σ(I): 0 / Redundancy: 5.8 % / Biso Wilson estimate: 15.2 Å2 / Rmerge(I) obs: 0.068 |
| Reflection shell | Resolution: 1.7→1.76 Å / Rmerge(I) obs: 0.32 / % possible all: 93 |
| Reflection | *PLUS Num. measured all: 248448 |
| Reflection shell | *PLUS % possible obs: 93 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1ROM Resolution: 1.7→50 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 16.4 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.7→1.81 Å / Rfactor Rfree error: 0.017 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.8 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 5.1 % / Rfactor obs: 0.195 / Rfactor Rfree: 0.25 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 16.4 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.297 / % reflection Rfree: 4.7 % / Rfactor Rwork: 0.276 |
