 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1byg: KINASE DOMAIN OF HUMAN C-TERMINAL SRC KINASE (CSK) IN COMPLEX WIT... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1byg | ||||||
|---|---|---|---|---|---|---|---|
| Title | KINASE DOMAIN OF HUMAN C-TERMINAL SRC KINASE (CSK) IN COMPLEX WITH INHIBITOR STAUROSPORINE | ||||||
 Components Components | PROTEIN (C-TERMINAL SRC KINASE) | ||||||
 Keywords Keywords | TRANSFERASE / PROTEIN KINASE / C-TERMINAL SRC KINASE / PHOSPHORYLATION / STAUROSPORINE | ||||||
| Function / homology |  Function and homology information Function and homology information[Src-family] C-terminal protein kinase / regulation of Fc receptor mediated stimulatory signaling pathway / negative regulation of Golgi to plasma membrane protein transport / adherens junction organization / negative regulation of bone resorption / proline-rich region binding / oligodendrocyte differentiation / negative regulation of low-density lipoprotein particle clearance / cellular response to peptide hormone stimulus / negative regulation of T cell activation ...[Src-family] C-terminal protein kinase / regulation of Fc receptor mediated stimulatory signaling pathway / negative regulation of Golgi to plasma membrane protein transport / adherens junction organization / negative regulation of bone resorption / proline-rich region binding / oligodendrocyte differentiation / negative regulation of low-density lipoprotein particle clearance / cellular response to peptide hormone stimulus / negative regulation of T cell activation / negative regulation of interleukin-6 production / negative regulation of phagocytosis / Phosphorylation of CD3 and TCR zeta chains / protein kinase A catalytic subunit binding / Co-inhibition by PD-1 / RHOH GTPase cycle / negative regulation of T cell receptor signaling pathway / GAB1 signalosome / T cell costimulation / Integrin signaling / Negative regulation of FLT3 / protein tyrosine kinase binding / non-membrane spanning protein tyrosine kinase activity / negative regulation of ERK1 and ERK2 cascade / Signaling by high-kinase activity BRAF mutants / MAP2K and MAPK activation / Signaling by RAF1 mutants / cell-cell junction / Signaling by moderate kinase activity BRAF mutants / Paradoxical activation of RAF signaling by kinase inactive BRAF / Signaling downstream of RAS mutants / Signaling by BRAF and RAF1 fusions / T cell receptor signaling pathway / protein tyrosine kinase activity / protein phosphatase binding / adaptive immune response / protein phosphorylation / intracellular signal transduction / negative regulation of cell population proliferation / extracellular exosome / ATP binding / metal ion binding / identical protein binding / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Antson, A.A. / Lamers, M.B.A.C. / Scott, R.K. / Williams, D.H. / Hubbard, R.E. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1999 Title: Structure of the protein tyrosine kinase domain of C-terminal Src kinase (CSK) in complex with staurosporine. Authors: Lamers, M.B. / Antson, A.A. / Hubbard, R.E. / Scott, R.K. / Williams, D.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1byg.cif.gz | 68.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1byg.ent.gz | 49.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1byg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/by/1bygftp://data.pdbj.org/pub/pdb/validation_reports/by/1byg | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1fmkS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj













- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 31181.918 Da / Num. of mol.: 1 / Fragment: KINASE DOMAIN Source method: isolated from a genetically manipulated source Details: THE C-TERMINAL HISTIDINE TAG WAS NOT OBSERVED IN THE DENSITY AND IS NOT PRESENT IN THE SEQRES. Source: (gene. exp.) Homo sapiens (human)Description: THE CDNA FOR THE CATALYTIC DOMAIN OF CSK WAS ISOLATED BY POLYMERASE CHAIN REACTION FROM GRANULOCYTE CDNA OBTAINED FROM HUMAN LUNG TISSUE. A HEXAHISTIDINE TAG WAS ENGINEERED AT THE C- ...Description: THE CDNA FOR THE CATALYTIC DOMAIN OF CSK WAS ISOLATED BY POLYMERASE CHAIN REACTION FROM GRANULOCYTE CDNA OBTAINED FROM HUMAN LUNG TISSUE. A HEXAHISTIDINE TAG WAS ENGINEERED AT THE C-TERMINUS TO AID PURIFICATION. Cell: GRANULOCYTE / Organ: LUNG / Plasmid: PQE60-CSK / Gene (production host): CSK / Production host:  |
|---|---|
| #2: Chemical | ChemComp-STU / 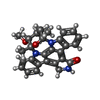  Mass: 466.531 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C28H26N4O3 / Comment: antibiotic*YM Mass: 466.531 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C28H26N4O3 / Comment: antibiotic*YM |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 152 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 152 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 38 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 9 Details: VAPOUR DIFFUSION, PROTEIN SOLUTION (10 MG/ML) CONTAINING 1MM STAUROSPORINE WAS MIXED IN 1:1 RATIO WITH THE RESERVOIR SOLUTION. THE RESERVOIR SOLUTION CONTAINED 0.2M MGCL2, 0.1M TRISHCL PH 9.0, 24% PEG 4000. | ||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Dec 15, 1997 / Details: MIRROR |
| Radiation | Monochromator: GRAPHITE CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→30 Å / Num. obs: 30746 / % possible obs: 93.6 % / Observed criterion σ(I): 0 / Redundancy: 3 % / Biso Wilson estimate: 36 Å2 / Rmerge(I) obs: 0.075 / Net I/σ(I): 13.7 |
| Reflection shell | Resolution: 2.4→2.49 Å / Redundancy: 1.7 % / Rmerge(I) obs: 0.224 / Mean I/σ(I) obs: 3.3 / % possible all: 61.8 |
| Reflection shell | *PLUS % possible obs: 61.8 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1FMK Resolution: 2.4→20 Å / SU B: 10.8 / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 43.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.4 Å / Lowest resolution: 20 Å / σ(F): 0 / % reflection Rfree: 10 % / Rfactor obs: 0.199 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 43.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
