 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1by5 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | FHUA FROM E. COLI, WITH ITS LIGAND FERRICHROME | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | METAL BINDING PROTEIN / FHUA / MEMBRANE PROTEIN / LIGAND-GATED / IRON TRANSPORT / FERRICHROME | |||||||||
| Function / homology |  Function and homology information Function and homology informationsiderophore-iron import into cell / siderophore transmembrane transport / siderophore uptake transmembrane transporter activity / virion binding / toxic substance binding / cell outer membrane / signaling receptor activity / intracellular iron ion homeostasis / iron ion binding / protein domain specific binding / membrane Similarity search - Function | |||||||||
| Biological species |   Ustilago sphaerogena (fungus) Ustilago sphaerogena (fungus) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.6 Å | |||||||||
 Authors Authors | Locher, K.P. / Rees, B. / Koebnik, R. / Mitschler, A. / Moulinier, L. / Rosenbusch, J.P. / Moras, D. | |||||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 1998 Title: Transmembrane signaling across the ligand-gated FhuA receptor: crystal structures of free and ferrichrome-bound states reveal allosteric changes. Authors: Locher, K.P. / Rees, B. / Koebnik, R. / Mitschler, A. / Moulinier, L. / Rosenbusch, J.P. / Moras, D. #1: Journal: Eur.J.Biochem. / Year: 1997Title: Oligomeric States and Siderophore-Binding of the Ligand-Gated Fhua-Protein Forming Channels Across E. Coli Outer Membranes Authors: Locher, K.P. / Rosenbusch, J.P. #2: Journal: J.Am.Chem.Soc. / Year: 1980Title: Crystal Structure of Ferrichrome and a Comparison with the Structure of Ferrichrome A Authors: Van Der Helm, D. / Baker, J.R. / Eng-Wilmot, D.L. / Hossain, M.B. / Loghry, R.A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1by5.cif.gz | 154.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1by5.ent.gz | 120.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1by5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/by/1by5ftp://data.pdbj.org/pub/pdb/validation_reports/by/1by5 | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 78929.016 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) | ||||||
|---|---|---|---|---|---|---|---|
| #2: Protein/peptide | Mass: 705.716 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Ustilago sphaerogena (fungus) | ||||||
| #3: Chemical | ChemComp-OES / 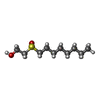  Mass: 206.345 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C10H22O2S Mass: 206.345 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C10H22O2S#4: Chemical | ChemComp-FE / |   Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 143 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 143 / Source method: isolated from a natural source / Formula: H2OCompound details | CHAIN B IS A CYCLIC PEPTIDE | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 4 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.32 Å3/Da / Density % sol: 63.1 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.2 / Details: pH 6.2 | ||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, sitting dropDetails: protein solution is mixed in a 1:2 ratio with well solution | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM14 / Wavelength: 0.9998 / Beamline: BM14 / Wavelength: 0.9998 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Aug 15, 1997 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9998 Å / Relative weight: 1 |
| Reflection | Resolution: 2.59→30 Å / Num. obs: 30766 / % possible obs: 92.8 % / Redundancy: 5.3 % / Biso Wilson estimate: 61.2 Å2 / Rmerge(I) obs: 0.06 |
| Reflection | *PLUS Rmerge(I) obs: 0.06 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.6→12 Å / Rfactor Rfree error: 0.006 / Data cutoff high rms absF: 1572333.99 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Stereochemistry target values: MAXIMUM LIKELYHOOD USING AMPLITUDES
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 56.1 Å2 / ksol: 0.34 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 53 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→12 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.6→2.74 Å / Rfactor Rfree error: 0.02 / Total num. of bins used: 7
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.4 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 5.1 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 53 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.288 / % reflection Rfree: 5.5 % / Rfactor Rwork: 0.259 |
