 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1bfd | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | BENZOYLFORMATE DECARBOXYLASE FROM PSEUDOMONAS PUTIDA | ||||||
 要素 要素 | BENZOYLFORMATE DECARBOXYLASE | ||||||
 キーワード キーワード | LYASE / CARBON-CARBON / DECARBOXYLASE / MANDELATE CATABOLISM / THIAMIN DIPHOSPHATE | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報benzoylformate decarboxylase / benzoylformate decarboxylase activity / mandelate catabolic process / acetolactate synthase activity / thiamine pyrophosphate binding / flavin adenine dinucleotide binding / magnesium ion binding 類似検索 - 分子機能 | ||||||
| 生物種 |  Pseudomonas putida (バクテリア) Pseudomonas putida (バクテリア) | ||||||
| 手法 |  X線回折 / 多重同系置換 / 解像度: 1.6 Å X線回折 / 多重同系置換 / 解像度: 1.6 Å | ||||||
 データ登録者 データ登録者 | Hasson, M.S. / Muscate, A. / Mcleish, M.J. / Polovnikova, L.S. / Gerlt, J.A. / Kenyon, G.L. / Petsko, G.A. / Ringe, D. | ||||||
 引用 引用 | ジャーナル: Biochemistry / 年: 1998 タイトル: The crystal structure of benzoylformate decarboxylase at 1.6 A resolution: diversity of catalytic residues in thiamin diphosphate-dependent enzymes. 著者: Hasson, M.S. / Muscate, A. / McLeish, M.J. / Polovnikova, L.S. / Gerlt, J.A. / Kenyon, G.L. / Petsko, G.A. / Ringe, D. #1: ジャーナル: Protein Sci. / 年: 1995タイトル: Purification and Crystallization of Benzoylformate Decarboxylase 著者: Hasson, M.S. / Muscate, A. / Henehan, G.T. / Guidinger, P.F. / Petsko, G.A. / Ringe, D. / Kenyon, G.L. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1bfd.cif.gz | 121.4 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1bfd.ent.gz | 92 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1bfd.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/bf/1bfdftp://data.pdbj.org/pub/pdb/validation_reports/bf/1bfd | HTTPS FTP |
|---|
-関連構造データ











-リンク
 PDBj
PDBj



- 集合体
集合体
| 登録構造単位 | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||
| 単位格子 |
| |||||||||||||||
| Components on special symmetry positions |
|
-要素
| #1: タンパク質 | 分子量: 56402.746 Da / 分子数: 1 / 由来タイプ: 組換発現 / 由来: (組換発現) Pseudomonas putida (バクテリア) / 株: RF4738 / 遺伝子: MDLC / プラスミド: PKK233-2 / 遺伝子 (発現宿主): MDLC / 発現宿主: | ||||||
|---|---|---|---|---|---|---|---|
| #2: 化合物 |   分子量: 40.078 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Ca 分子量: 40.078 Da / 分子数: 2 / 由来タイプ: 合成 / 式: Ca#3: 化合物 | ChemComp-MG / |   分子量: 24.305 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Mg 分子量: 24.305 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Mg#4: 化合物 | ChemComp-TPP / | 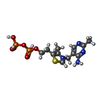  分子量: 425.314 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C12H19N4O7P2S 分子量: 425.314 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C12H19N4O7P2S#5: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 346 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 346 / 由来タイプ: 天然 / 式: H2O |
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.44 Å3/Da / 溶媒含有率: 50 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 8.5 詳細: CRYSTALS WERE GROWN AT ROOM TEMPERATURE BY HANGING-DROP VAPOR DIFFUSION AGAINST A WELL SOLUTION OF 22% (V/V) POLYETHYLENE GLYCOL WITH AN AVERAGE MOLECULAR WEIGHT OF 400 KDA (PEG 400), 0.15 M ...詳細: CRYSTALS WERE GROWN AT ROOM TEMPERATURE BY HANGING-DROP VAPOR DIFFUSION AGAINST A WELL SOLUTION OF 22% (V/V) POLYETHYLENE GLYCOL WITH AN AVERAGE MOLECULAR WEIGHT OF 400 KDA (PEG 400), 0.15 M CACL2, 0.5% (V/V) MPD, 0.1 M TRISCL (PH 8.5). DROPS CONTAINED EQUAL VOLUMES (2 MICROL) OF WELL SOLUTION AND PURIFIED BENZOYLFORMATE DECARBOXYLASE [10 MG/ML IN 0.1 MM MGCL2, 0.2 MM TDP, 25 MM NAHEPES (PH 7.0)]., vapor diffusion - hanging drop PH範囲: 7.0-8.5 / Temp details: room temp | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 手法: 蒸気拡散法, ハンギングドロップ法詳細: drop solution was mixed with an equal volume of reservoir solution | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 277 K |
|---|---|
| 放射光源 | 由来: 回転陽極 / タイプ: RIGAKU RUH2R / 波長: 1.5418 |
| 検出器 | タイプ: RIGAKU RAXIS IIC / 検出器: IMAGE PLATE / 日付: 1994年3月1日 / 詳細: COLLIMATOR |
| 放射 | モノクロメーター: NI FILTER / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.5418 Å / 相対比: 1 |
| 反射 | 最高解像度: 1.6 Å / Num. obs: 59179 / % possible obs: 81 % / Observed criterion σ(I): 1 / 冗長度: 3.7 % / Biso Wilson estimate: 17.4 Å2 / Rmerge(I) obs: 0.051 / Net I/σ(I): 14 |
| 反射 シェル | 解像度: 1.6→1.7 Å / Mean I/σ(I) obs: 2.2 / % possible all: 32 |
| 反射 | *PLUS Num. measured all: 217078 |
| 反射 シェル | *PLUS % possible obs: 32 % |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 多重同系置換 / 解像度: 1.6→5 Å / Rfactor Rfree error: 0.003 / Data cutoff high absF: 1000000 / Data cutoff low absF: 0.1 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 1 詳細: VAL 524 IS THE LAST RESIDUE FOR WHICH INTERPRETABLE ELECTRON DENSITY WAS PRESENT.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 18.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 1.6→5 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 1.6→1.7 Å / Rfactor Rfree error: 0.018 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: X-PLOR / バージョン: 3 / 分類: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS Num. reflection obs: 59179 / Rfactor obs: 0.15 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | *PLUS Rfactor obs: 0.275 |
