 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1bcj: MANNOSE-BINDING PROTEIN-A MUTANT (QPDWGHV) COMPLEXED WITH N-ACETY... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1bcj | ||||||
|---|---|---|---|---|---|---|---|
| Title | MANNOSE-BINDING PROTEIN-A MUTANT (QPDWGHV) COMPLEXED WITH N-ACETYL-D-GALACTOSAMINE | ||||||
 Components Components | MANNOSE-BINDING PROTEIN-A | ||||||
 Keywords Keywords | LECTIN / C-TYPE LECTIN / CALCIUM-BINDING PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationcalcium-dependent carbohydrate binding / complement activation, lectin pathway / oligosaccharide binding / : / surfactant homeostasis / phosphatidylinositol-4-phosphate binding / protein homotrimerization / D-mannose binding / polysaccharide binding / complement activation, classical pathway ...calcium-dependent carbohydrate binding / complement activation, lectin pathway / oligosaccharide binding / : / surfactant homeostasis / phosphatidylinositol-4-phosphate binding / protein homotrimerization / D-mannose binding / polysaccharide binding / complement activation, classical pathway / multivesicular body / positive regulation of phagocytosis / calcium-dependent protein binding / protease binding / defense response to Gram-positive bacterium / calcium ion binding / protein homodimerization activity / : / identical protein binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Kolatkar, A.R. / Weis, W.I. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 1998 Title: Mechanism of N-acetylgalactosamine binding to a C-type animal lectin carbohydrate-recognition domain. Authors: Kolatkar, A.R. / Leung, A.K. / Isecke, R. / Brossmer, R. / Drickamer, K. / Weis, W.I. #1: Journal: J.Biol.Chem. / Year: 1996Title: Selective Sugar Binding to the Carbohydrate Recognition Domains of the Rat Hepatic and Macrophage Asialoglycoprotein Receptors Authors: Iobst, S.T. / Drickamer, K. #2: Journal: J.Biol.Chem. / Year: 1996Title: Structural Basis of Galactose Recognition by C-Type Animal Lectins Authors: Kolatkar, A.R. / Weis, W.I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1bcj.cif.gz | 113.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1bcj.ent.gz | 86.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1bcj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bc/1bcjftp://data.pdbj.org/pub/pdb/validation_reports/bc/1bcj | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1bchC  1afbS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |















-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 17074.391 Da / Num. of mol.: 3 / Fragment: CLOSTRIPAIN FRAGMENT RESIDUES 73 - 226 Mutation: S154V, E185Q, N187D, H189W, G190Y, S191G, INS (H192, G193, L194, G195, G196), T202H Source method: isolated from a genetically manipulated source Source: (gene. exp.) Description: THE BACTERIALLY EXPRESSED MATERIAL WAS DIGESTED WITH CLOSTRIPAIN TO PRODUCE THE PROTEIN USED IN THE CRYSTAL STRUCTURE ANALYSIS Plasmid: PINIIIOMPA2 / Production host:  #2: Sugar | 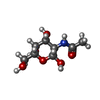  Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 3 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 3Source method: isolated from a genetically manipulated source Formula: C8H15NO6 #3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: Ca#4: Chemical |   Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 319 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 319 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.2 Å3/Da / Density % sol: 60.9 % Description: 20% 2-METHYL,2-4 PENTANEDIOL (CRYOPROTECTANT), 200MM N-ACETYL-D-GALACTOSAMINE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8 Details: 12% PEG8000, 100MM TRIS/PH8.0, 20MM CALCIUM CHLORIDE, 10MM SODIUM CHLORIDE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / Method: vapor diffusion, hanging dropDetails: drop solution was mixed with an equal volume of reservoir solution | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE / Date: Jul 1, 1997 |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→30 Å / Num. obs: 35683 / % possible obs: 95.7 % / Observed criterion σ(I): -3 / Redundancy: 2.5 % / Biso Wilson estimate: 23.3 Å2 / Rmerge(I) obs: 0.057 / Rsym value: 0.057 / Net I/σ(I): 11.4 |
| Reflection shell | Resolution: 2.1→2.2 Å / Redundancy: 2.4 % / Rmerge(I) obs: 0.283 / Mean I/σ(I) obs: 3.8 / Rsym value: 0.283 / % possible all: 96.9 |
| Reflection shell | *PLUS % possible obs: 96.9 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1AFB Resolution: 2.1→30 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 2643.491 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Stereochemistry target values: MAXIMUM LIKELIHOOD USING AMPLITUDES
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 42.5 Å2 / ksol: 0.32 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 36.4 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.2 Å / Rfactor Rfree error: 0.014 / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.3C / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor obs: 0.258 |
