 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1bbz: CRYSTAL STRUCTURE OF THE ABL-SH3 DOMAIN COMPLEXED WITH A DESIGNED... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1bbz | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE ABL-SH3 DOMAIN COMPLEXED WITH A DESIGNED HIGH-AFFINITY PEPTIDE LIGAND: IMPLICATIONS FOR SH3-LIGAND INTERACTIONS | ||||||
 Components Components |
| ||||||
 Keywords Keywords | COMPLEX (TRANSFERASE/PEPTIDE) / COMPLEX (TRANSFERASE-PEPTIDE) / SIGNAL TRANSDUCTION / SH3 DOMAIN / COMPLEX (TRANSFERASE-PEPTIDE) complex | ||||||
| Function / homology |  Function and homology information Function and homology informationprotein localization to cytoplasmic microtubule plus-end / DNA conformation change / response to epinephrine / phospholipase C-inhibiting G protein-coupled receptor signaling pathway / negative regulation of ubiquitin-protein transferase activity / podocyte apoptotic process / regulation of postsynaptic specialization assembly / positive regulation of phospholipase C/protein kinase C signal transduction / regulation of modification of synaptic structure / nicotinate-nucleotide adenylyltransferase activity ...protein localization to cytoplasmic microtubule plus-end / DNA conformation change / response to epinephrine / phospholipase C-inhibiting G protein-coupled receptor signaling pathway / negative regulation of ubiquitin-protein transferase activity / podocyte apoptotic process / regulation of postsynaptic specialization assembly / positive regulation of phospholipase C/protein kinase C signal transduction / regulation of modification of synaptic structure / nicotinate-nucleotide adenylyltransferase activity / delta-catenin binding / Role of ABL in ROBO-SLIT signaling / mitochondrial depolarization / positive regulation of extracellular matrix organization / neuropilin signaling pathway / neuropilin binding / regulation of cell motility / bubble DNA binding / positive regulation of establishment of T cell polarity / cellular response to dopamine / positive regulation of blood vessel branching / proline-rich region binding / mitogen-activated protein kinase binding / regulation of Cdc42 protein signal transduction / regulation of hematopoietic stem cell differentiation / syntaxin binding / positive regulation of dendrite development / regulation of axon extension / regulation of T cell differentiation / positive regulation of cell migration involved in sprouting angiogenesis / Myogenesis / HDR through Single Strand Annealing (SSA) / platelet-derived growth factor receptor-beta signaling pathway / RUNX2 regulates osteoblast differentiation / Fc-gamma receptor signaling pathway involved in phagocytosis / vascular endothelial cell response to oscillatory fluid shear stress / regulation of endocytosis / regulation of microtubule polymerization / negative regulation of long-term synaptic potentiation / myoblast proliferation / associative learning / cardiac muscle cell proliferation / positive regulation of focal adhesion assembly / actin monomer binding / ephrin receptor signaling pathway / positive regulation of vasoconstriction / RHO GTPases Activate WASPs and WAVEs / cellular response to transforming growth factor beta stimulus / positive regulation of T cell migration / endothelial cell migration / negative regulation of double-strand break repair via homologous recombination / regulation of cell adhesion / mismatch repair / ephrin receptor binding / four-way junction DNA binding / ruffle / positive regulation of stress fiber assembly / signal transduction in response to DNA damage / phosphotyrosine residue binding / actin filament polymerization / positive regulation of substrate adhesion-dependent cell spreading / positive regulation of endothelial cell migration / SH2 domain binding / response to endoplasmic reticulum stress / Turbulent (oscillatory, disturbed) flow shear stress activates signaling by PIEZO1 and integrins in endothelial cells / integrin-mediated signaling pathway / protein kinase C binding / protein modification process / regulation of autophagy / protein serine/threonine kinase activator activity / regulation of actin cytoskeleton organization / non-specific protein-tyrosine kinase / FCGR3A-mediated phagocytosis / non-membrane spanning protein tyrosine kinase activity / Regulation of actin dynamics for phagocytic cup formation / intrinsic apoptotic signaling pathway in response to DNA damage / autophagy / positive regulation of fibroblast proliferation / cellular response to hydrogen peroxide / epidermal growth factor receptor signaling pathway / enzyme activator activity / sequence-specific double-stranded DNA binding / kinase activity / Cyclin D associated events in G1 / actin filament binding / positive regulation of neuron apoptotic process / manganese ion binding / mitotic cell cycle / actin cytoskeleton / Recruitment and ATM-mediated phosphorylation of repair and signaling proteins at DNA double strand breaks / positive regulation of cytosolic calcium ion concentration / Factors involved in megakaryocyte development and platelet production / MLL4 and MLL3 complexes regulate expression of PPARG target genes in adipogenesis and hepatic steatosis / growth cone / RUNX1 regulates transcription of genes involved in differentiation of HSCs / actin cytoskeleton organization / cellular response to oxidative stress / protein tyrosine kinase activity / response to oxidative stress / nuclear membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.65 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.65 Å | ||||||
 Authors Authors | Pisabarro, M.T. / Serrano, L. / Wilmanns, M. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1998 Title: Crystal structure of the abl-SH3 domain complexed with a designed high-affinity peptide ligand: implications for SH3-ligand interactions. Authors: Pisabarro, M.T. / Serrano, L. / Wilmanns, M. #1: Journal: Biochemistry / Year: 1996Title: Rational Design of Specific High-Affinity Peptide Ligands for the Abl-SH3 Domain Authors: Pisabarro, M.T. / Serrano, L. #2: Journal: Nat.Struct.Biol. / Year: 1994Title: High-Resolution Crystal Structures of Tyrosine Kinase SH3 Domains Complexed with Proline-Rich Peptides Authors: Musacchio, A. / Saraste, M. / Wilmanns, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1bbz.cif.gz | 72.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1bbz.ent.gz | 54.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1bbz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bb/1bbzftp://data.pdbj.org/pub/pdb/validation_reports/bb/1bbz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1aboS S: Starting model for refinement |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 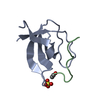
| ||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||
| 3 | 
| ||||||||||||||||||||||||||||
| 4 | 
| ||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 6423.080 Da / Num. of mol.: 4 / Fragment: SH3 DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host:  #2: Protein/peptide | Mass: 1035.149 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source #3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 269 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 269 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.31 Å3/Da / Density % sol: 46.68 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion / pH: 3.1 Details: CRYSTALS WITH DIMENSIONS 0.25X0.25X0.25 MM3 WERE OBTAINED AT ROOM TEMPERATURE BY VAPOUR DIFFUSION AGAINST A RESERVOIR CONTAINING 0.1 M CITRIC ACID PH 3.1, 2 M AMMONIUM SULPHATE, 0.2 M SODIUM ...Details: CRYSTALS WITH DIMENSIONS 0.25X0.25X0.25 MM3 WERE OBTAINED AT ROOM TEMPERATURE BY VAPOUR DIFFUSION AGAINST A RESERVOIR CONTAINING 0.1 M CITRIC ACID PH 3.1, 2 M AMMONIUM SULPHATE, 0.2 M SODIUM CHLORIDE, AND 1MM DTT/EDTA. THE HANGING DROP CONTAINED 1:1 RATIO OF RESERVOIR AND PROTEIN-PEPTIDE SOLUTIONS., vapor diffusion | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: BW7B / Wavelength: 0.88 / Beamline: BW7B / Wavelength: 0.88 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Oct 1, 1996 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.88 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→39.3 Å / Num. obs: 226846 / % possible obs: 99.7 % / Observed criterion σ(I): 0 / Redundancy: 6.59 % / Biso Wilson estimate: 15.4 Å2 / Rmerge(I) obs: 0.058 / Rsym value: 0.058 / Net I/σ(I): 15.1 |
| Reflection shell | Resolution: 1.6→1.63 Å / Redundancy: 3.7 % / Rmerge(I) obs: 0.58 / Mean I/σ(I) obs: 1.9 / Rsym value: 0.58 / % possible all: 98.4 |
| Reflection | *PLUS Num. obs: 34430 / Num. measured all: 226846 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1ABO Resolution: 1.65→8 Å / Cross valid method: FREE R-FACTOR / σ(F): 1
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 13.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.65→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
