 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1av6: VACCINIA METHYLTRANSFERASE VP39 COMPLEXED WITH M7G CAPPED RNA HEX... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1av6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | VACCINIA METHYLTRANSFERASE VP39 COMPLEXED WITH M7G CAPPED RNA HEXAMER AND S-ADENOSYLHOMOCYSTEINE | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE/RNA / SINGLE-STRANDED RNA / METHYLTRANSFERASE / RNA CAP / POLY(A) POLYMERASE / VACCINIA / MRNA PROCESSING / TRANSCRIPTION / COMPLEX (TRANSFERASE-RNA) / TRANSFERASE-RNA complex | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of mRNA 3'-end processing / 7-methylguanosine mRNA capping / virion component / methylation / methyltransferase cap1 / methyltransferase cap1 activity / DNA-templated transcription / RNA binding Similarity search - Function | ||||||
| Biological species |  Vaccinia virus WR Vaccinia virus WR | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR / Resolution: 2.8 Å X-RAY DIFFRACTION / MOLECULAR / Resolution: 2.8 Å | ||||||
 Authors Authors | Hodel, A.E. / Gershon, P.D. / Quiocho, F.A. | ||||||
 Citation Citation | Journal: Mol.Cell / Year: 1998 Title: Structural basis for sequence-nonspecific recognition of 5'-capped mRNA by a cap-modifying enzyme. Authors: Hodel, A.E. / Gershon, P.D. / Quiocho, F.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1av6.cif.gz | 77.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1av6.ent.gz | 61.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1av6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/av/1av6ftp://data.pdbj.org/pub/pdb/validation_reports/av/1av6 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1vptS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: RNA chain | Mass: 1946.277 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source |
|---|---|
| #2: Protein | Mass: 34573.008 Da / Num. of mol.: 1 / Mutation: C-TERMINAL DELETION OF 26 RESIDUES Source method: isolated from a genetically manipulated source Source: (gene. exp.) Vaccinia virus WR / Genus: Orthopoxvirus / Strain: Western Reserve / Gene: PAPS, VACWR095, F9 / Species (production host): Escherichia coli / Production host:  Keywords: MUTATION:C-TERMINAL DELETION OF 26 RESIDUES / References: UniProt: P07617, methyltransferase cap1 Keywords: MUTATION:C-TERMINAL DELETION OF 26 RESIDUES / References: UniProt: P07617, methyltransferase cap1 |
| #3: Chemical | ChemComp-MGT / 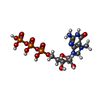  Mass: 539.223 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H20N5O14P3 Mass: 539.223 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H20N5O14P3 |
| #4: Chemical | ChemComp-SAH /   Type: L-peptide linking / Mass: 384.411 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H20N6O5S Type: L-peptide linking / Mass: 384.411 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H20N6O5S |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.77 Å3/Da / Density % sol: 56 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8.5 / Details: pH 8.50 | ||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop / Details: used to seeding / pH: 7 | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 103 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 |
| Detector | Type: SIEMENS / Detector: AREA DETECTOR / Date: Aug 15, 1997 / Details: GOBEL MIRROR |
| Radiation | Monochromator: NI FILTER / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 2.7→20 Å / Num. obs: 9755 / % possible obs: 92 % / Observed criterion σ(I): 2 / Redundancy: 3 % / Biso Wilson estimate: 27 Å2 / Rmerge(I) obs: 0.095 / Net I/σ(I): 20 |
| Reflection shell | Resolution: 2.7→2.8 Å / Redundancy: 2 % / Rmerge(I) obs: 0.21 / Mean I/σ(I) obs: 6 |
| Reflection | *PLUS Highest resolution: 2.7 Å / Lowest resolution: 20 Å / % possible obs: 92 % / Observed criterion σ(I): 2 / Redundancy: 3 % / Num. measured all: 22144 / Biso Wilson estimate: 27 Å2 |
| Reflection shell | *PLUS Highest resolution: 2.7 Å / Lowest resolution: 2.8 Å / Redundancy: 2 % / Mean I/σ(I) obs: 6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR Starting model: PDB ENTRY 1VPT Resolution: 2.8→8 Å / Data cutoff high absF: 100000 / Data cutoff low absF: 0.0001 / Isotropic thermal model: GROUP / Cross valid method: FREE-R / σ(F): 2 Details: SINGLE PHOSPHATE LINKS BETWEEN RESIDUES ARE PHOSPHOROTHIOATE, I.E., M7GPPPG-PS-A-PS-A-PS-A-PS-A-PS-A. PHOSPHOROTHIOLATES ARE LEFT AS PHOSPHATE IN THIS INITIAL DEPOSITION.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 13 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.8 Å / Lowest resolution: 8 Å / σ(F): 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 13 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
