| 登録情報 | データベース: PDB / ID: 1anb
|
|---|
| タイトル | ANIONIC TRYPSIN MUTANT WITH SER 214 REPLACED BY GLU |
|---|
 要素 要素 | ANIONIC TRYPSIN |
|---|
 キーワード キーワード | SERINE PROTEASE / TRYPSIN / ANIONIC / HYDROLASE |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
Antimicrobial peptides / Alpha-defensins / Activation of Matrix Metalloproteinases / Neutrophil degranulation / collagen catabolic process / trypsin / digestion / response to nutrient / serine-type endopeptidase activity / calcium ion binding ...Antimicrobial peptides / Alpha-defensins / Activation of Matrix Metalloproteinases / Neutrophil degranulation / collagen catabolic process / trypsin / digestion / response to nutrient / serine-type endopeptidase activity / calcium ion binding / proteolysis / : / extracellular region類似検索 - 分子機能 : / Serine proteases, trypsin family, histidine active site / Serine proteases, trypsin family, serine active site / Serine proteases, trypsin family, histidine active site. / Serine proteases, trypsin family, serine active site. / Peptidase S1A, chymotrypsin family / Serine proteases, trypsin domain profile. / Trypsin-like serine protease / Serine proteases, trypsin domain / Trypsin ...: / Serine proteases, trypsin family, histidine active site / Serine proteases, trypsin family, serine active site / Serine proteases, trypsin family, histidine active site. / Serine proteases, trypsin family, serine active site. / Peptidase S1A, chymotrypsin family / Serine proteases, trypsin domain profile. / Trypsin-like serine protease / Serine proteases, trypsin domain / Trypsin / Trypsin-like serine proteases / Thrombin, subunit H / Peptidase S1, PA clan, chymotrypsin-like fold / Peptidase S1, PA clan / Beta Barrel / Mainly Beta類似検索 - ドメイン・相同性 |
|---|
| 生物種 |   Rattus rattus (エジプトネズミ) Rattus rattus (エジプトネズミ) |
|---|
| 手法 |  X線回折 / DIFFERENCE FOURIER / 解像度: 2.8 Å X線回折 / DIFFERENCE FOURIER / 解像度: 2.8 Å |
|---|
 データ登録者 データ登録者 | Fletterick, R.J. / Mcgrath, M.E. |
|---|
 引用 引用 | ジャーナル: Biochemistry / 年: 1992
タイトル: Perturbing the polar environment of Asp102 in trypsin: consequences of replacing conserved Ser214.
著者: McGrath, M.E. / Vasquez, J.R. / Craik, C.S. / Yang, A.S. / Honig, B. / Fletterick, R.J. |
|---|
| 履歴 | | 登録 | 1994年12月21日 | 処理サイト: BNL |
|---|
| 改定 1.0 | 1997年4月1日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2008年3月24日 | Group: Version format compliance |
|---|
| 改定 1.2 | 2011年7月13日 | Group: Version format compliance |
|---|
| 改定 1.3 | 2021年11月3日 | Group: Database references / Derived calculations / Other
カテゴリ: database_2 / pdbx_database_status ...database_2 / pdbx_database_status / struct_conn / struct_ref_seq_dif / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.process_site / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
| 改定 1.4 | 2024年10月9日 | Group: Data collection / Structure summary
カテゴリ: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / pdbx_entry_details / pdbx_modification_feature |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード










 PDBj
PDBj







 集合体
集合体



 分子量: 40.078 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Ca
分子量: 40.078 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Ca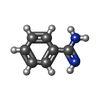
 分子量: 120.152 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C7H8N2
分子量: 120.152 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C7H8N2 分子量: 18.015 Da / 分子数: 149 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 149 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析