 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ajh | ||||||
|---|---|---|---|---|---|---|---|
| Title | PHOTOPRODUCT OF CARBONMONOXY MYOGLOBIN AT 40 K | ||||||
 Components Components | MYOGLOBIN | ||||||
 Keywords Keywords | OXYGEN TRANSPORT / RESPIRATORY PROTEIN / HEME / PHOTOPRODUCT INTERMEDIATE | ||||||
| Function / homology |  Function and homology information Function and homology informationOxidoreductases; Acting on other nitrogenous compounds as donors / nitrite reductase activity / sarcoplasm / Oxidoreductases; Acting on a peroxide as acceptor; Peroxidases / removal of superoxide radicals / oxygen carrier activity / peroxidase activity / oxygen binding / heme binding / extracellular exosome / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / DIFFERENCE FOURIER / Resolution: 1.69 Å X-RAY DIFFRACTION / SYNCHROTRON / DIFFERENCE FOURIER / Resolution: 1.69 Å | ||||||
 Authors Authors | Teng, T.Y. / Srajer, V. / Moffat, K. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1994 Title: Photolysis-induced structural changes in single crystals of carbonmonoxy myoglobin at 40 K. Authors: Teng, T.Y. / Srajer, V. / Moffat, K. #1: Journal: Biochemistry / Year: 1997Title: Initial Trajectory of Carbon Monoxide After Photodissociation from Myoglobin at Cryogenic Temperatures Authors: Teng, T.Y. / Srajer, V. / Moffat, K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ajh.cif.gz | 49.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ajh.ent.gz | 34.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1ajh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/aj/1ajhftp://data.pdbj.org/pub/pdb/validation_reports/aj/1ajh | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ajgC 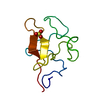 1krnS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 17234.951 Da / Num. of mol.: 1 / Source method: isolated from a natural source Details: CARBONMONOXY MYOGLOBIN, PHOTODISSOCIATED CO MOLECULE IS NOT BOUND TO THE HEME, HEME BOUND TO NE2 OF HIS 93 Source: (natural) | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-HEM / |   Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4#4: Chemical | ChemComp-CMO / | 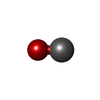  Mass: 28.010 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CO Mass: 28.010 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CO#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 183 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 183 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.84 Å3/Da / Density % sol: 33.17 % Description: THE CRYSTAL WAS ILLUMINATED BY A HE-NE LASER FOR 90 SECONDS BEFORE EACH X-RAY EXPOSURE. DATA WERE COLLECTED IN DARK USING AN OPEN FLOW NITROGEN/HELIUM CRYOSTAT FOR COOLING. |
|---|---|
| Crystal grow | pH: 6 Details: AS DESCRIBED IN KENDREW,J.C. AND PARRISH,R.G., PROC. ROY. SOC. A (LONDON) 238, 305-324 (1956), pH 6.0 |
| Crystal grow | *PLUS Method: unknownDetails: Kendrew, J.C., (1956) Proc. Royal Soc. A, 238, 305. |
-Data collection
| Diffraction | Mean temperature: 40 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X26C / Wavelength: 1.123 / Beamline: X26C / Wavelength: 1.123 |
| Detector | Type: FUJI / Detector: IMAGE PLATE / Date: Jun 30, 1993 / Details: YES |
| Radiation | Monochromator: YES / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.123 Å / Relative weight: 1 |
| Reflection | Resolution: 1.69→27.5 Å / Num. obs: 13618 / % possible obs: 95.8 % / Observed criterion σ(I): 0 / Redundancy: 3.3 % / Rmerge(I) obs: 0.05 / Net I/σ(I): 10.9 |
| Reflection shell | Resolution: 1.69→1.77 Å / Redundancy: 2.7 % / Rmerge(I) obs: 0.146 / Mean I/σ(I) obs: 5 / % possible all: 83.5 |
| Reflection | *PLUS Num. measured all: 51604 / Rmerge(I) obs: 0.05 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: DIFFERENCE FOURIER Starting model: PDB ENTRY 1KRN Resolution: 1.69→10 Å / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / σ(F): 2 Details: THE FOLLOWING RESIDUES HAVE BEEN MODELED WITH TWO CONFORMERS -- 4 GLU, 21 VAL, 26 GLN, 31 ARG, 61 LEU.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.16 Å / Luzzati d res low obs: 10 Å / Luzzati sigma a obs: 0.08 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.69→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.69→1.77 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
