 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1abw | ||||||
|---|---|---|---|---|---|---|---|
| Title | DEOXY RHB1.1 (RECOMBINANT HEMOGLOBIN) | ||||||
 Components Components | (HEMOGLOBIN-BASED BLOOD ...) x 2 | ||||||
 Keywords Keywords | OXYGEN TRANSPORT / HEME / RESPIRATORY PROTEIN / ERYTHROCYTE | ||||||
| Function / homology |  Function and homology information Function and homology informationcellular oxidant detoxification / Heme assimilation / nitric oxide transport / hemoglobin alpha binding / hemoglobin binding / haptoglobin-hemoglobin complex / renal absorption / hemoglobin complex / oxygen transport / Scavenging of heme from plasma ...cellular oxidant detoxification / Heme assimilation / nitric oxide transport / hemoglobin alpha binding / hemoglobin binding / haptoglobin-hemoglobin complex / renal absorption / hemoglobin complex / oxygen transport / Scavenging of heme from plasma / erythrocyte development / endocytic vesicle lumen / blood vessel diameter maintenance / hydrogen peroxide catabolic process / oxygen carrier activity / carbon dioxide transport / response to hydrogen peroxide / Heme signaling / Erythrocytes take up oxygen and release carbon dioxide / Erythrocytes take up carbon dioxide and release oxygen / Cytoprotection by HMOX1 / oxygen binding / Late endosomal microautophagy / platelet aggregation / regulation of blood pressure / Chaperone Mediated Autophagy / positive regulation of nitric oxide biosynthetic process / tertiary granule lumen / Factors involved in megakaryocyte development and platelet production / blood microparticle / ficolin-1-rich granule lumen / iron ion binding / inflammatory response / heme binding / Neutrophil degranulation / : / extracellular exosome / extracellular region / membrane / metal ion binding / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Kundrot, C.E. / Kroeger, K.S. | ||||||
 Citation Citation | Journal: Structure / Year: 1997 Title: Structures of a hemoglobin-based blood substitute: insights into the function of allosteric proteins. Authors: Kroeger, K.S. / Kundrot, C.E. #1: Journal: Nature / Year: 1992Title: A Human Recombinant Haemoglobin Designed for Use as a Blood Substitute Authors: Looker, D. / Abbott-Brown, D. / Cozart, P. / Durfee, S. / Hoffman, S. / Mathews, A.J. / Miller-Roehrich, J. / Shoemaker, S. / Trimble, S. / Fermi, G. / Komiyama, N.H. / Nagai, K. / Stetler, G.L. | ||||||
| History |
| ||||||
| Remark 7 | A NOTE FROM THE AUTHOR: RESIDUES 1001-1002 and 2001-2002 MODEL LOW OCCUPANCY STRUCTURES THAT HAVE ... A NOTE FROM THE AUTHOR: RESIDUES 1001-1002 and 2001-2002 MODEL LOW OCCUPANCY STRUCTURES THAT HAVE THEIR AMINO TERMINUS IN REGISTER WITH VAL A143. THE SPATIAL ARRANGEMENT OF TWO ALPHA CHAINS OF NORMAL HEMOGLOBIN CAN BE REPRESENTED SCHEMATICALLY AS GHIJ F A EDCB WHERE FGHIJ IS ONE COVALENT CHAIN AND ABCDE THE OTHER COVALENT CHAIN. IN THIS MUTANT HEMOGLOBIN, THERE IS A COVALENT BOND BETWEEN RESIDUE J AND A, BUT NOT BETWEEN E AND F. IN THE CRYSTAL, THE AUTHORS COULD HAVE GHIJ BCDE F A OR A F EDCB JIHG (1) (2) THE "F-A" CONTACT WOULD BE LIKE NORMAL HEMOGLOBIN, BUT THE "E-F" CONTACT HAS A COVALENT LINKAGE THAT BREAKS THE SYMMETRY. THE ONLY WAY TO TELL IF THE CRYSTAL STRUCTURE IS (1) OR (2) OR A MIXTURE OF THE TWO IS TO LOOK AT THE "E-F" AND "J-A" REGIONS. IN THIS STRUCTURE, IT COULD BE MOSTLY (1), BUT ABOUT 20% OF THE MOLECULES MAY BE FLIPPED OVER LIKE (2). THAT FLIPPING WOULD PUT THE N-TERMINAL METHIONINE ON TOP OF VAL 143. THUS, THE ELECTRON DENSITY MAP AT POSITION VAL 143 IS MOSTLY FROM VAL 143, BUT IT ALSO CONTAINS A LITTLE ELECTRON DENSITY FROM MET 1 FROM FLIPPED MOLECULES. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1abw.cif.gz | 137.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1abw.ent.gz | 107.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1abw.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ab/1abwftp://data.pdbj.org/pub/pdb/validation_reports/ab/1abw | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1abyC  2hhbS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj















- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-HEMOGLOBIN-BASED BLOOD ... , 2 types, 3 molecules ABD
| #1: Protein | Mass: 30371.846 Da / Num. of mol.: 1 Mutation: CHAIN A IS COMPOSED OF TWO COPIES OF HEMOGLOBIN ALPHA CHAIN JOINED BY AN ADDITIONAL RESIDUE, GLY 142 Source method: isolated from a genetically manipulated source Details: HUMAN HEMOGLOBIN HAS TWO ALPHA CHAINS WHICH ARE CALLED CHAINS A AND C IN OTHER PDB FILES. IN THIS ENTRY THE TWO ALPHA CHAINS HAVE BEEN COVALENTLY JOINED TOGETHER BY ONE GLYCINE RESIDUE TO ...Details: HUMAN HEMOGLOBIN HAS TWO ALPHA CHAINS WHICH ARE CALLED CHAINS A AND C IN OTHER PDB FILES. IN THIS ENTRY THE TWO ALPHA CHAINS HAVE BEEN COVALENTLY JOINED TOGETHER BY ONE GLYCINE RESIDUE TO FORM ONE COVALENTLY LINKED POLYPEPTIDE CHAIN. Source: (gene. exp.) Homo sapiens (human) / Organ: BLOOD / Production host:  |
|---|---|
| #2: Protein | Mass: 15937.343 Da / Num. of mol.: 2 Mutation: CHAIN A IS COMPOSED OF TWO COPIES OF HEMOGLOBIN ALPHA CHAIN JOINED BY AN ADDITIONAL RESIDUE, GLY 142 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Organ: BLOOD / Production host: |
-Non-polymers , 4 types, 381 molecules 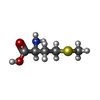



| #3: Chemical |  Type: L-peptide linking / Mass: 149.211 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H11NO2S Type: L-peptide linking / Mass: 149.211 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H11NO2S#4: Chemical |  Type: L-peptide linking / Mass: 131.173 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H13NO2 Type: L-peptide linking / Mass: 131.173 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H13NO2#5: Chemical | ChemComp-HEM /  Mass: 616.487 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C34H32FeN4O4#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 373 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Compound details | HUMAN HEMOGLOBIN HAS TWO ALPHA CHAINS WHICH ARE CALLED CHAINS A AND C IN OTHER PDB FILES. IN THIS ...HUMAN HEMOGLOBIN |
|---|---|
| Nonpolymer details | THE RESIDUES 1001-1002 AND 2001-2002 REPRESENT LOW OCCUPANCY DIPEPTIDES (MET-LEU) THAT HAVE THEIR ...THE RESIDUES 1001-1002 AND 2001-2002 REPRESENT LOW OCCUPANCY DIPEPTIDES |
| Sequence details | GLY A 142 COVALENTLY |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 5 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.19 Å3/Da / Density % sol: 43.75 % | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.2 / Details: pH 6.2 | ||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 5 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 295 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: SIEMENS / Detector: AREA DETECTOR / Date: Mar 1, 1993 / Details: COLLIMATOR |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→82 Å / Num. obs: 34054 / % possible obs: 77.1 % / Observed criterion σ(I): 2 / Redundancy: 2.1 % / Rmerge(I) obs: 0.046 |
| Reflection shell | Resolution: 2→2.2 Å / % possible all: 56.1 |
| Reflection | *PLUS Num. measured all: 70840 |
| Reflection shell | *PLUS % possible obs: 56.1 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2HHB Resolution: 2→5 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 100000 / Data cutoff low absF: 0.1 / Cross valid method: THROUGHOUT / σ(F): 2 Details: A NOTE FROM THE AUTHOR: CHAINS L AND P MODEL LOW OCCUPANCY STRUCTURES THAT HAVE THEIR AMINO TERMINUS IN REGISTER WITH VAL A143. THE SPATIAL ARRANGEMENT OF TWO ALPHA CHAINS OF NORMAL ...Details: A NOTE FROM THE AUTHOR: CHAINS L AND P MODEL LOW OCCUPANCY STRUCTURES THAT HAVE THEIR AMINO TERMINUS IN REGISTER WITH VAL A143. THE SPATIAL ARRANGEMENT OF TWO ALPHA CHAINS OF NORMAL HEMOGLOBIN CAN BE REPRESENTED SCHEMATICALLY AS GHIJ F A EDCB WHERE FGHIJ IS ONE COVALENT CHAIN AND ABCDE THE OTHER COVALENT CHAIN. IN THIS MUTANT HEMOGLOBIN, THERE IS A COVALENT BOND BETWEEN RESIDUE J AND A, BUT NOT BETWEEN E AND F. IN THE CRYSTAL, THE AUTHORS COULD HAVE GHIJ BCDE F A OR A F EDCB JIHG (1) (2) THE "F-A" CONTACT WOULD BE LIKE NORMAL HEMOGLOBIN, BUT THE "E-F" CONTACT HAS A COVALENT LINKAGE THAT BREAKS THE SYMMETRY. THE ONLY WAY TO TELL IF THE CRYSTAL STRUCTURE IS (1) OR (2) OR A MIXTURE OF THE TWO IS TO LOOK AT THE "E-F" AND "J-A" REGIONS. IN THIS STRUCTURE, IT COULD BE MOSTLY (1), BUT ABOUT 20% OF THE MOLECULES MAY BE FLIPPED OVER LIKE (2). THAT FLIPPING WOULD PUT THE N-TERMINAL METHIONINE ON TOP OF VAL 143. THUS, THE ELECTRON DENSITY MAP AT POSITION VAL 143 IS MOSTLY FROM VAL 143, BUT IT ALSO CONTAINS A LITTLE ELECTRON DENSITY FROM MET 1 FROM FLIPPED MOLECULES.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 13.3 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→5 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.09 Å / Rfactor Rfree error: 0.014 / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor obs: 0.213 |
