 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1a6g | ||||||
|---|---|---|---|---|---|---|---|
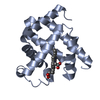
| Title | CARBONMONOXY-MYOGLOBIN, ATOMIC RESOLUTION | ||||||
 Components Components | MYOGLOBIN | ||||||
 Keywords Keywords | HEME PROTEIN / MODEL COMPOUNDS / OXYGEN STORAGE / LIGAND BINDING GEOMETRY / CONFORMATIONAL SUBSTATES | ||||||
| Function / homology |  Function and homology information Function and homology informationOxidoreductases; Acting on other nitrogenous compounds as donors / nitrite reductase activity / sarcoplasm / Oxidoreductases; Acting on a peroxide as acceptor; Peroxidases / removal of superoxide radicals / oxygen carrier activity / peroxidase activity / oxygen binding / heme binding / extracellular exosome / metal ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.15 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.15 Å | ||||||
 Authors Authors | Vojtechovsky, J. / Chu, K. / Berendzen, J. / Sweet, R.M. / Schlichting, I. | ||||||
 Citation Citation | Journal: Biophys.J. / Year: 1999 Title: Crystal structures of myoglobin-ligand complexes at near-atomic resolution. Authors: Vojtechovsky, J. / Chu, K. / Berendzen, J. / Sweet, R.M. / Schlichting, I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1a6g.cif.gz | 91 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1a6g.ent.gz | 69.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1a6g.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1a6g_validation.pdf.gz | 808.7 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1a6g_full_validation.pdf.gz | 815.5 KB | Display | |
| Data in XML | 1a6g_validation.xml.gz | 11.6 KB | Display | |
| Data in CIF | 1a6g_validation.cif.gz | 16.6 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a6/1a6gftp://data.pdbj.org/pub/pdb/validation_reports/a6/1a6g | HTTPS FTP |
-Related structure data
| Related structure data |  1a6kC  1a6mC  1a6nC  1mbcS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |


















-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 17048.787 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-HEM / |   Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4#4: Chemical | ChemComp-CMO / | 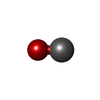  Mass: 28.010 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CO Mass: 28.010 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CO#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 184 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 184 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.8 Å3/Da / Density % sol: 35.18 % | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6 Details: PROTEIN WAS CRYSTALLIZED FROM AMMONIUM SULPHATE, 50MM POTASSIUM PHOSPHATE, PH 6.0 | |||||||||||||||
| Crystal | *PLUS | |||||||||||||||
| Crystal grow | *PLUS pH: 7 / Method: batch method | |||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X12C / Wavelength: 0.91 / Beamline: X12C / Wavelength: 0.91 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE AREA DETECTOR / Date: Jan 1, 1997 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.91 Å / Relative weight: 1 |
| Reflection | Resolution: 1.15→50 Å / Num. obs: 42857 / % possible obs: 91 % / Rsym value: 0.059 / Net I/σ(I): 20 |
| Reflection shell | Resolution: 1.15→1.2 Å / Mean I/σ(I) obs: 4 / Rsym value: 0.299 / % possible all: 63 |
| Reflection | *PLUS Num. measured all: 413744 / Rmerge(I) obs: 0.059 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1MBC Resolution: 1.15→8 Å / Num. parameters: 12713 / Num. restraintsaints: 19055 / Cross valid method: FREE R StereochEM target val spec case: HEME - PARAMETERS BASED ON CSD Stereochemistry target values: ENGH & HUBER Details: NO GEOMETRIC RESTRAINTS APPLIED TO IRON AND THE PLANAR ATOMS OF THE HEME. DATA CUTOFF -3.0 (SIGMA(I)) BAYESIAN DIFFERENCE REFINEMENT WAS USED AT THE FINAL STEP. SEE TERWILLIGER AND ...Details: NO GEOMETRIC RESTRAINTS APPLIED TO IRON AND THE PLANAR ATOMS OF THE HEME. DATA CUTOFF -3.0 (SIGMA(I)) BAYESIAN DIFFERENCE REFINEMENT WAS USED AT THE FINAL STEP. SEE TERWILLIGER AND BERENDZEN, ACTA CRYST. D52:1004-1011. THE SOLVENT MOLECULES 129,130,132,139 AND 146 CAN BE MODELED FOR ONE ALTERNATIVE PROTEIN CONFORMATION.
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 26 / Occupancy sum hydrogen: 1263.1 / Occupancy sum non hydrogen: 1415.9 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.15→8 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
