 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-9223: Cryo-EM structures and dynamics of substrate-engaged human 26S pr... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-9223 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome | |||||||||
 Map data Map data | State EA1 map of substrate-engaged human proteasome low pass-filtered to 3 Angstrom, without amplitude correction | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.0 Å | |||||||||
 Authors Authors | Mao YD | |||||||||
 Citation Citation | Journal: Nature / Year: 2019 Title: Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Authors: Yuanchen Dong / Shuwen Zhang / Zhaolong Wu / Xuemei Li / Wei Li Wang / Yanan Zhu / Svetla Stoilova-McPhie / Ying Lu / Daniel Finley / Youdong Mao /   Abstract: The proteasome is an ATP-dependent, 2.5-megadalton molecular machine that is responsible for selective protein degradation in eukaryotic cells. Here we present cryo-electron microscopy structures of ...The proteasome is an ATP-dependent, 2.5-megadalton molecular machine that is responsible for selective protein degradation in eukaryotic cells. Here we present cryo-electron microscopy structures of the substrate-engaged human proteasome in seven conformational states at 2.8-3.6 Å resolution, captured during breakdown of a polyubiquitylated protein. These structures illuminate a spatiotemporal continuum of dynamic substrate-proteasome interactions from ubiquitin recognition to substrate translocation, during which ATP hydrolysis sequentially navigates through all six ATPases. There are three principal modes of coordinated hydrolysis, featuring hydrolytic events in two oppositely positioned ATPases, in two adjacent ATPases and in one ATPase at a time. These hydrolytic modes regulate deubiquitylation, initiation of translocation and processive unfolding of substrates, respectively. Hydrolysis of ATP powers a hinge-like motion in each ATPase that regulates its substrate interaction. Synchronization of ATP binding, ADP release and ATP hydrolysis in three adjacent ATPases drives rigid-body rotations of substrate-bound ATPases that are propagated unidirectionally in the ATPase ring and unfold the substrate. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_9223.map.gz | 749 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-9223-v30.xmlemd-9223.xml | 15.1 KB 15.1 KB | Display Display | EMDB header |
| Images | 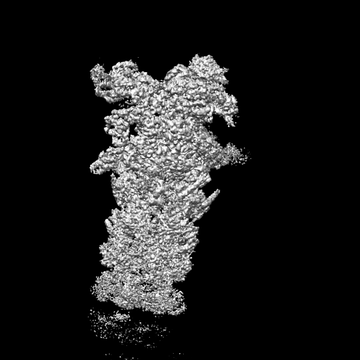 emd_9223.png emd_9223.png | 30.2 KB | ||
| Others | emd_9223_additional_1.map.gzemd_9223_additional_2.map.gz | 729.3 MB 757.7 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-9223ftp://ftp.pdbj.org/pub/emdb/structures/EMD-9223 http://ftp.pdbj.org/pub/emdb/structures/EMD-9223ftp://ftp.pdbj.org/pub/emdb/structures/EMD-9223 | HTTPS FTP |
-Related structure data
| Related structure data |  9215C  9216C  9217C  9218C  9219C  9220C  9221C  9222C  9224C  9225C  9226C  9227C  9228C  9229C  6msbC  6msdC  6mseC  6msgC  6mshC  6msjC  6mskC C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10669 (Title: Cryo-EM dataset of the substrate-engaged human 26S proteasome Data size: 13.9 TB Data #1: Drift-corrected frame-averaged super-counting mode micrographs and extracted particles of substrate-engaged human 26S proteasome [micrographs - single frame]) |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_9223.map.gz / Format: CCP4 / Size: 824 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | State EA1 map of substrate-engaged human proteasome low pass-filtered to 3 Angstrom, without amplitude correction | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
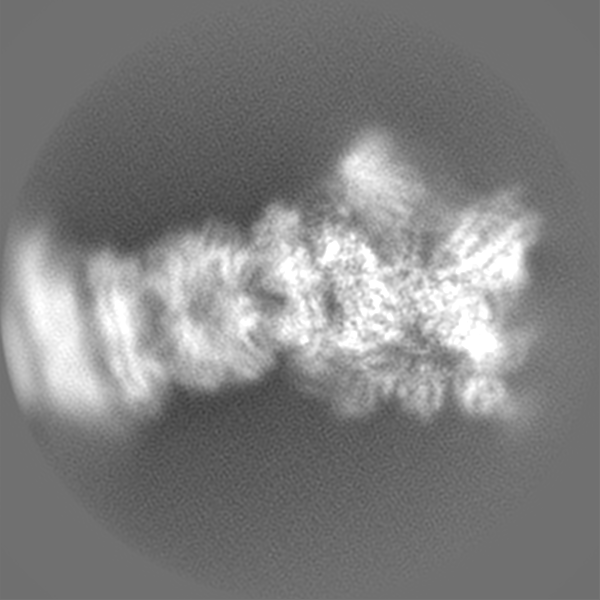
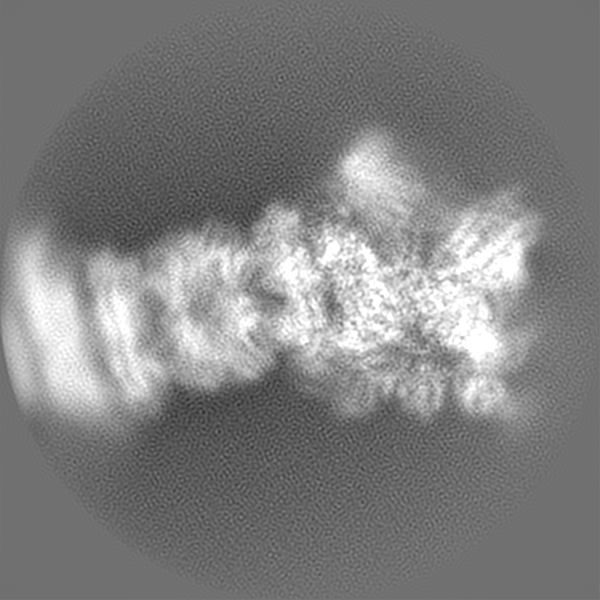
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.685 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
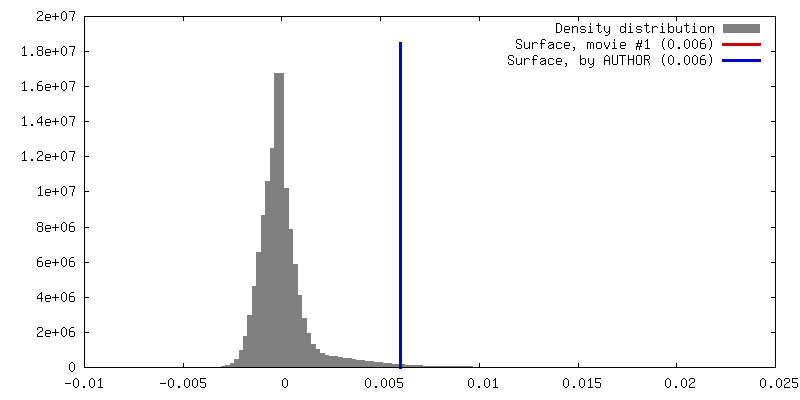
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)


















-Supplemental data
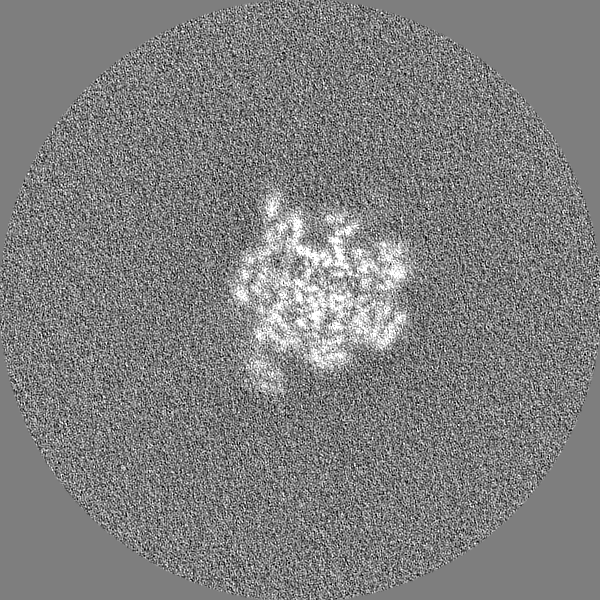
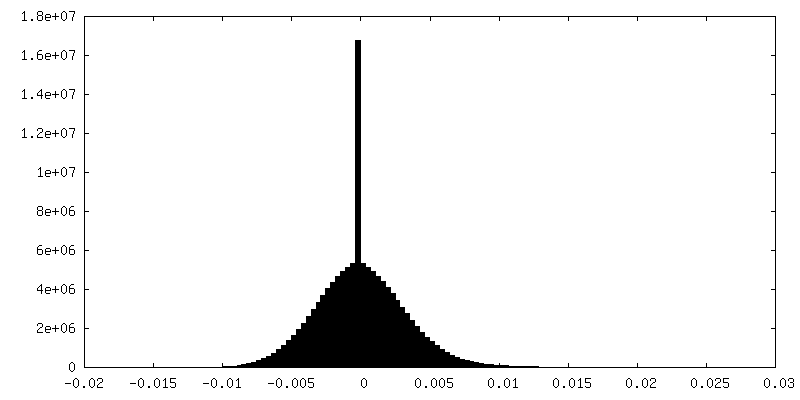
-Additional map: Unfiltered, uncorrected map of state EA1
| File | emd_9223_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unfiltered, uncorrected map of state EA1 | ||||||||||||
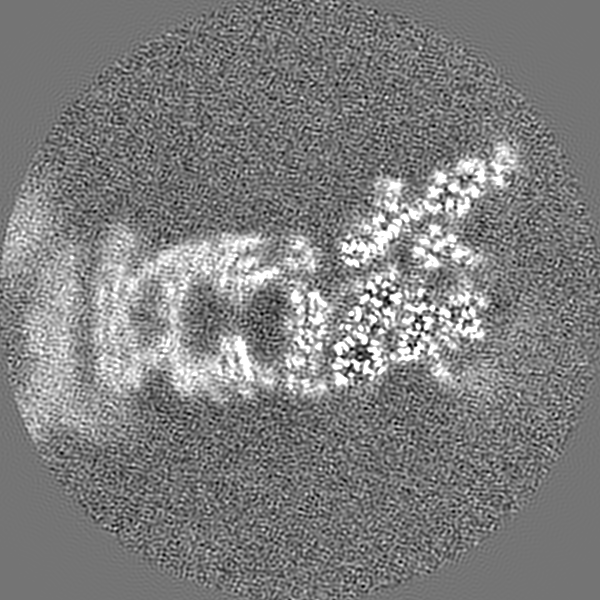
| Projections & Slices |
| ||||||||||||
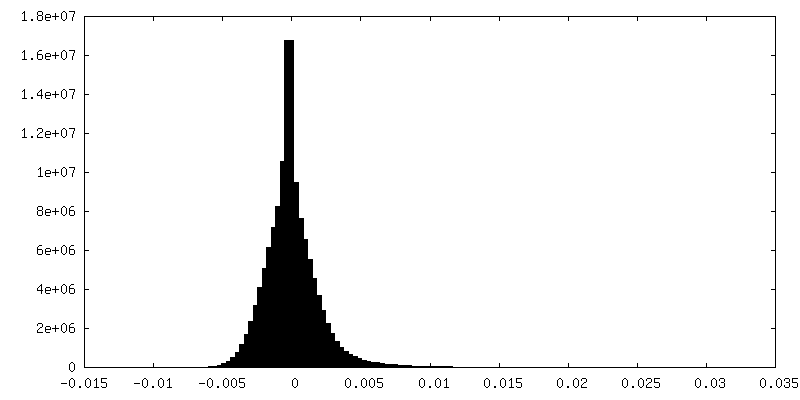
| Density Histograms |

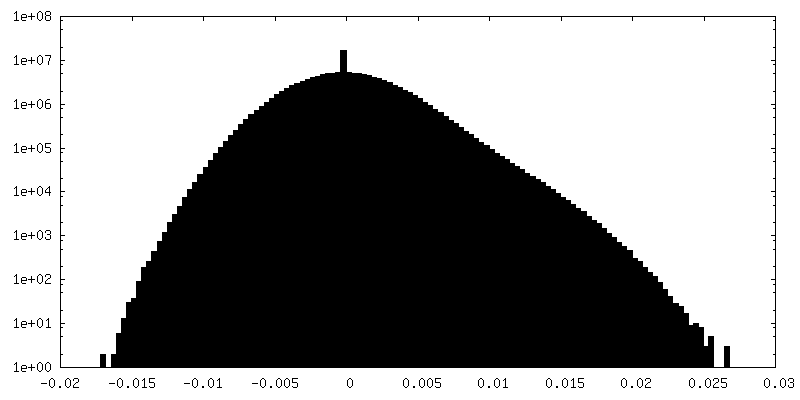
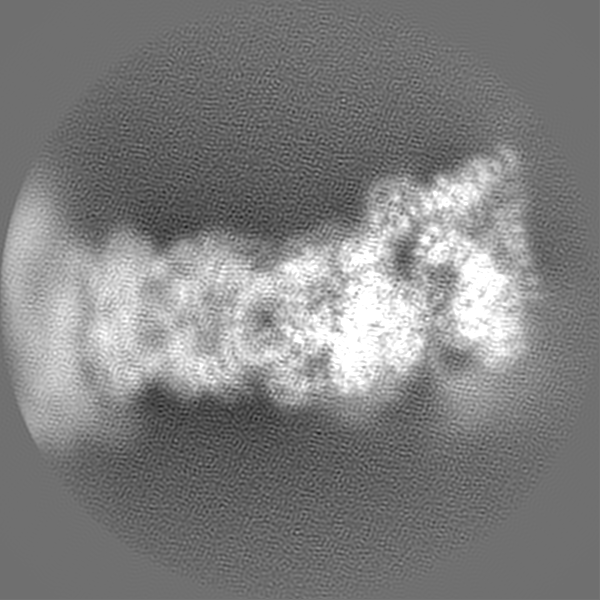
-Additional map: State EA1 map of substrate-engaged human proteasome low...
| File | emd_9223_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | State EA1 map of substrate-engaged human proteasome low pass-filtered to 3 Angstrom, with amplitude correction with a B-factor of -40 | ||||||||||||
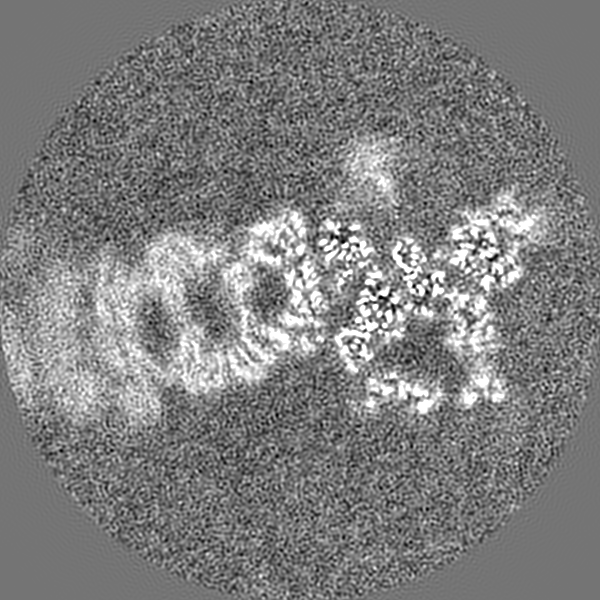
| Projections & Slices |
| ||||||||||||
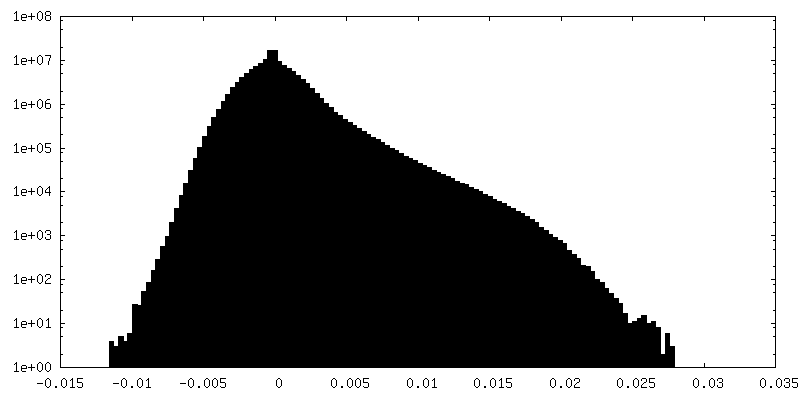
| Density Histograms |

- Sample components
Sample components
-Entire : Proteasome
| Entire | Name: Proteasome |
|---|---|
| Components |
|
-Supramolecule #1: Proteasome
| Supramolecule | Name: Proteasome / type: complex / ID: 1 / Parent: 0 |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Recombinant expression | Organism: Homo sapiens (human) |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.5 |
|---|---|
| Grid | Details: unspecified |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Average electron dose: 44.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Final reconstruction | Applied symmetry - Point group: C1 (asymmetric) / Resolution.type: BY AUTHOR / Resolution: 3.0 Å / Resolution method: FSC 0.143 CUT-OFF / Number images used: 105157 |
|---|---|
| Initial angle assignment | Type: COMMON LINE |
| Final angle assignment | Type: PROJECTION MATCHING |
