Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-8543: 3D negative stain EM structure of Pom152, the major component of ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-8543 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | 3D negative stain EM structure of Pom152, the major component of the membrane ring of the nuclear pore complex | |||||||||
 Map data Map data | 3D negative stain EM structure of Pom152 | |||||||||
 Sample Sample |
| |||||||||
| Function / homology |  Function and homology information Function and homology informationnuclear pore transmembrane ring / spindle pole body duplication / nuclear pore organization / structural constituent of nuclear pore / nucleocytoplasmic transport / nuclear envelope lumen / nuclear pore / mRNA transport / protein-membrane adaptor activity / nuclear periphery ...nuclear pore transmembrane ring / spindle pole body duplication / nuclear pore organization / structural constituent of nuclear pore / nucleocytoplasmic transport / nuclear envelope lumen / nuclear pore / mRNA transport / protein-membrane adaptor activity / nuclear periphery / cell periphery / protein import into nucleus / nuclear envelope / nuclear membrane / mitochondrion Similarity search - Function | |||||||||
| Biological species |  | |||||||||
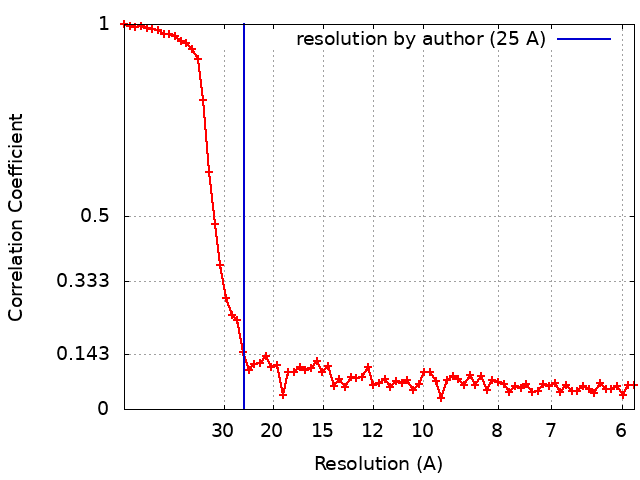
| Method | single particle reconstruction / negative staining / Resolution: 25.0 Å | |||||||||
 Authors Authors | Upla P / Kim SJ / Sampathkumar P / Dutta K / Cahill SM / Chemmama IE / Williams R / Bonanno JB / Rice WJ / Stokes DL ...Upla P / Kim SJ / Sampathkumar P / Dutta K / Cahill SM / Chemmama IE / Williams R / Bonanno JB / Rice WJ / Stokes DL / Cowburn D / Almo SC / Sali A / Rout MP / Fernandez-Martinez J | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: Structure / Year: 2017 Title: Molecular Architecture of the Major Membrane Ring Component of the Nuclear Pore Complex. Authors: Paula Upla / Seung Joong Kim / Parthasarathy Sampathkumar / Kaushik Dutta / Sean M Cahill / Ilan E Chemmama / Rosemary Williams / Jeffrey B Bonanno / William J Rice / David L Stokes / David ...Authors: Paula Upla / Seung Joong Kim / Parthasarathy Sampathkumar / Kaushik Dutta / Sean M Cahill / Ilan E Chemmama / Rosemary Williams / Jeffrey B Bonanno / William J Rice / David L Stokes / David Cowburn / Steven C Almo / Andrej Sali / Michael P Rout / Javier Fernandez-Martinez / Abstract: The membrane ring that equatorially circumscribes the nuclear pore complex (NPC) in the perinuclear lumen of the nuclear envelope is composed largely of Pom152 in yeast and its ortholog Nup210 (or ...The membrane ring that equatorially circumscribes the nuclear pore complex (NPC) in the perinuclear lumen of the nuclear envelope is composed largely of Pom152 in yeast and its ortholog Nup210 (or Gp210) in vertebrates. Here, we have used a combination of negative-stain electron microscopy, nuclear magnetic resonance, and small-angle X-ray scattering methods to determine an integrative structure of the ∼120 kDa luminal domain of Pom152. Our structural analysis reveals that the luminal domain is formed by a flexible string-of-pearls arrangement of nine repetitive cadherin-like Ig-like domains, indicating an evolutionary connection between NPCs and the cell adhesion machinery. The 16 copies of Pom152 known to be present in the yeast NPC are long enough to form the observed membrane ring, suggesting how interactions between Pom152 molecules help establish and maintain the NPC architecture. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_8543.map.gz | 17.1 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-8543-v30.xmlemd-8543.xml | 16.7 KB 16.7 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_8543_fsc.xml | 7.6 KB | Display | FSC data file |
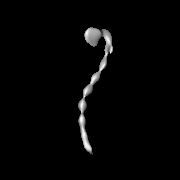
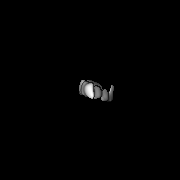
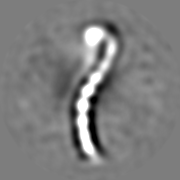
| Images |  emd_8543.png emd_8543.png | 13 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-8543ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8543 http://ftp.pdbj.org/pub/emdb/structures/EMD-8543ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8543 | HTTPS FTP |
-Related structure data


-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_8543.map.gz / Format: CCP4 / Size: 22.2 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | 3D negative stain EM structure of Pom152 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 2.93 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
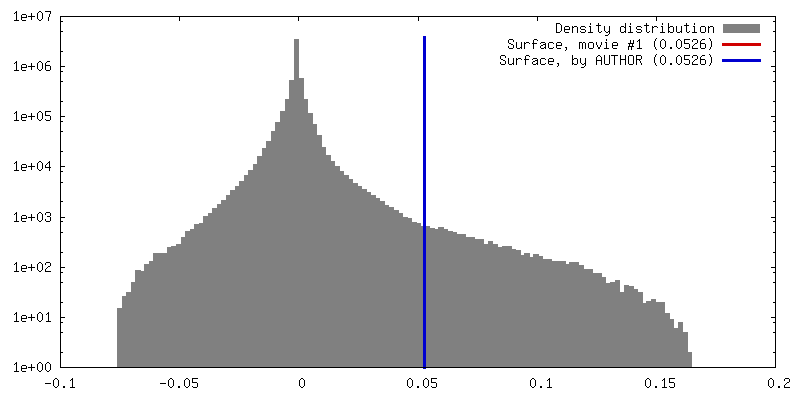
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)

















-Supplemental data
- Sample components
Sample components
-Entire : Nucleoporin Pom152
| Entire | Name: Nucleoporin Pom152 |
|---|---|
| Components |
|
-Supramolecule #1: Nucleoporin Pom152
| Supramolecule | Name: Nucleoporin Pom152 / type: cell / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: |
-Macromolecule #1: Pom152
| Macromolecule | Name: Pom152 / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Sequence | String: MEHRYNVFND TPRGNHWMGS SVSGSPRPSY SSRPNVNTTR RFQYSDDEPA EKIRPLRSRS FKSTESNIS DEKSRISERD SKDRYINGDK KVDIYSLPLI STDVLEISKQ RTFAVILFLI I QCYKIYDL VILKSGLPLS GLLFKNYRFN FISKYFIIDS FFLYVLPSFN ...String: MEHRYNVFND TPRGNHWMGS SVSGSPRPSY SSRPNVNTTR RFQYSDDEPA EKIRPLRSRS FKSTESNIS DEKSRISERD SKDRYINGDK KVDIYSLPLI STDVLEISKQ RTFAVILFLI I QCYKIYDL VILKSGLPLS GLLFKNYRFN FISKYFIIDS FFLYVLPSFN IPRLTFKPWV VY LQILAML LLNIFISSDH EFVLISLIMT TWRKLYTKEL SVTGSAINHH RIFDSSAHFK GAL TIKILP ENTAMFNPLH ESYCLPMDTN LFKINSIDVP IRINSTEEIE YIELEYRDLY TNSV ELRSL SKKDFKIIDN PKSFLKKDQS VLKSHSNDFE EGSTIRYLAV TLQDIGFYQI KKIVD SKKL NLKIHQSHLV VPYCPIASIT GTGSNDRCIG DSDNVSFEIQ GVPPMKLAYS KIVNGQ TFS YVDSSLQPEY FESPLQSSKS KQSFTQGELN DLKWGRNQPV NINLDSSITQ DGKFAYK ID KITDGLGNVV DFTSLPEELK KRYDLSYNFN VHEVPRAALE ERFDPKSPTK RSIAIVFE E IKNWISDIPY VISLSYTDAQ DKSKKIMNVT TDSLTKVLQA DLPGSYNLEY IESKFCPGE IVGKSNVLVT MPVAPTMEVK SFPILDQCVG QVGLNFELSF TGAPPYYYNT KIYKLENGER KLYDAKRYT SEGTRNRFSY SPPKEGNYEI VFDTVSNKLF TEPIKLEPVK EYTFKTSMRV K PSASLKLH HDLKLCLGDH SSVPVALKGQ GPFTLTYDII ETFSSKRKTF EIKEIKTNEY VI KTPVFTT GGDYILSLVS IKDSTGCVVG LSQPDAKIQV RRDIPSAAFN FFEPIKEAKI KHG SVTEIP LKLSGEGPFT VKFKHMDYDG NIVKEFENKF QNSYKPALKV SKEGLYQLVD IRDS SCQGN VIYRNSLYKV SFLEKPKFAI QDNHHITKVT ENLFSKEEVC QGMEGTVDLA LFGSP PFIL EYDLMAPNGH ISTKKIQVAT KYASLKLPNQ IPGEYITTIK AIFDGNYGES DIHFRE HQS ELIIKQTVHP IPDVAFADGG KTLRACAANV DQISFLEPIN LKFLQGESPF SITFSVY HE STSRTDQYTI DNIDSENFSF EKLYEGMKLG NHAITIDSVV DANGCVNSLI SGPRNQIL V SITDAPKIHI LDPSTEYCVG DYVAYQLNGV APFMIKYEFN GIPLKSKERS SQFVRLASE PGIISITSLQ DSSSQCIVDF TNPKLKSEFD DLSLNIHPIP SVTVSQGNYV TEDIREGDQA EVIFSFEGT PPFSLTYVRT EETDGKHGKR RSQVVETHKV TDIYSHEYKV ITSLQGTYEA I EITDAYCF AKNDLFFNN |
-Experimental details
-Structure determination
| Method | negative staining |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.4 Component:
Details: Solutions were made fresh from concentrated, and sterile filtered. | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Staining | Type: NEGATIVE / Material: Uranyl formate Details: 3 ul of protein was added to grids. The excess sample was blotted and the grid washed several times with water. The sample was stained with several drops of uranyl formate, blotted between ...Details: 3 ul of protein was added to grids. The excess sample was blotted and the grid washed several times with water. The sample was stained with several drops of uranyl formate, blotted between each stain. After the last staining (30 sec), the sample was blotted and air dried. | |||||||||||||||||||||
| Grid | Model: Electron Microscopy Sciences / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: CONTINUOUS / Pretreatment - Type: PLASMA CLEANING / Pretreatment - Atmosphere: OTHER | |||||||||||||||||||||
| Details | The endogenous sample was purified using affinity tags, then natively eluted and isolated on a 5-20% sucrose gradient. Fractions identified by SDS-PAGE and mass spectrometry. |
- Electron microscopy
Electron microscopy
| Microscope | JEOL 2100F |
|---|---|
| Image recording | Film or detector model: TVIPS TEMCAM-F224 (2k x 2k) / Number grids imaged: 2 / Number real images: 1220 / Average exposure time: 2.0 sec. / Average electron dose: 15.0 e/Å2 Details: Images were collected at tilt angles 0 and 40 degrees |
| Electron beam | Acceleration voltage: 120 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 40.0 µm / Calibrated magnification: 81911 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.0 mm / Nominal defocus max: 1.5 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 50000 |
| Sample stage | Specimen holder model: JEOL |