 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-7063: Cryo-electron microscopy structure of porcine delta coronavirus s... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-7063 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
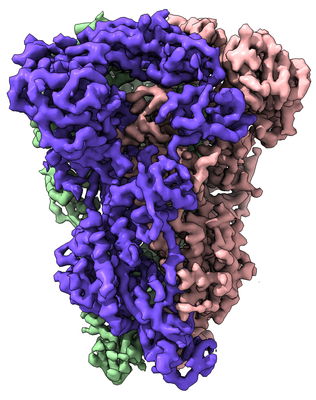
| Title | Cryo-electron microscopy structure of porcine delta coronavirus spike protein in the pre-fusion state | |||||||||
 Map data Map data | Delta coronavirus spike protein in the pre-fusion state | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | delta coronavirus / spike / pre-fusion / cryo-EM / VIRAL PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationreceptor-mediated virion attachment to host cell / endocytosis involved in viral entry into host cell / fusion of virus membrane with host plasma membrane / fusion of virus membrane with host endosome membrane / viral envelope / virion membrane / membrane Similarity search - Function | |||||||||
| Biological species |  Deltacoronavirus PDCoV/USA/Ohio137/2014 Deltacoronavirus PDCoV/USA/Ohio137/2014 | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.3 Å | |||||||||
 Authors Authors | Shang J / Zheng Y / Yang Y / Liu C / Geng Q / Tai W / Du L / Zhou Y / Zhang W / Li F | |||||||||
| Funding support |  United States, 2 items United States, 2 items
| |||||||||
 Citation Citation | Journal: J Virol / Year: 2018 Title: Cryo-Electron Microscopy Structure of Porcine Deltacoronavirus Spike Protein in the Prefusion State. Authors: Jian Shang / Yuan Zheng / Yang Yang / Chang Liu / Qibin Geng / Wanbo Tai / Lanying Du / Yusen Zhou / Wei Zhang / Fang Li /  Abstract: Coronavirus spike proteins from different genera are divergent, although they all mediate coronavirus entry into cells by binding to host receptors and fusing viral and cell membranes. Here, we ...Coronavirus spike proteins from different genera are divergent, although they all mediate coronavirus entry into cells by binding to host receptors and fusing viral and cell membranes. Here, we determined the cryo-electron microscopy structure of porcine deltacoronavirus (PdCoV) spike protein at 3.3-Å resolution. The trimeric protein contains three receptor-binding S1 subunits that tightly pack into a crown-like structure and three membrane fusion S2 subunits that form a stalk. Each S1 subunit contains two domains, an N-terminal domain (S1-NTD) and C-terminal domain (S1-CTD). PdCoV S1-NTD has the same structural fold as alpha- and betacoronavirus S1-NTDs as well as host galectins, and it recognizes sugar as its potential receptor. PdCoV S1-CTD has the same structural fold as alphacoronavirus S1-CTDs, but its structure differs from that of betacoronavirus S1-CTDs. PdCoV S1-CTD binds to an unidentified receptor on host cell surfaces. PdCoV S2 is locked in the prefusion conformation by structural restraint of S1 from a different monomeric subunit. PdCoV spike possesses several structural features that may facilitate immune evasion by the virus, such as its compact structure, concealed receptor-binding sites, and shielded critical epitopes. Overall, this study reveals that deltacoronavirus spikes are structurally and evolutionally more closely related to alphacoronavirus spikes than to betacoronavirus spikes; it also has implications for the receptor recognition, membrane fusion, and immune evasion by deltacoronaviruses as well as coronaviruses in general. IMPORTANCE In this study, we determined the cryo-electron microscopy structure of porcine deltacoronavirus (PdCoV) spike protein at a 3.3-Å resolution. This is the first atomic structure of a spike protein from the deltacoronavirus genus, which is divergent in amino acid sequences from the well-studied alpha- and betacoronavirus spike proteins. Here, we described the overall structure of the PdCoV spike and the detailed structure of each of its structural elements. Moreover, we analyzed the functions of each of the structural elements. Based on the structures and functions of these structural elements, we discussed the evolution of PdCoV spike protein in relation to the spike proteins from other coronavirus genera. This study combines the structure, function, and evolution of PdCoV spike protein and provides many insights into its receptor recognition, membrane fusion, and immune evasion. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_7063.map.gz | 60 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-7063-v30.xmlemd-7063.xml | 19.5 KB 19.5 KB | Display Display | EMDB header |
| Images |  emd_7063.png emd_7063.png | 169.7 KB | ||
| Filedesc metadata | emd-7063.cif.gz | 7.3 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-7063ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7063 http://ftp.pdbj.org/pub/emdb/structures/EMD-7063ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7063 | HTTPS FTP |
-Related structure data
| Related structure data |  6b7nMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_7063.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Delta coronavirus spike protein in the pre-fusion state | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.3 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Porcine delta coronavirus spike trimer in the pre-fusion state
| Entire | Name: Porcine delta coronavirus spike trimer in the pre-fusion state |
|---|---|
| Components |
|
-Supramolecule #1: Porcine delta coronavirus spike trimer in the pre-fusion state
| Supramolecule | Name: Porcine delta coronavirus spike trimer in the pre-fusion state type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: Deltacoronavirus PDCoV/USA/Ohio137/2014 |
-Macromolecule #1: Spike protein
| Macromolecule | Name: Spike protein / type: protein_or_peptide / ID: 1 / Number of copies: 3 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Deltacoronavirus PDCoV/USA/Ohio137/2014 |
| Molecular weight | Theoretical: 122.277266 KDa |
| Recombinant expression | Organism:   Spodoptera frugiperda (fall armyworm) Spodoptera frugiperda (fall armyworm) |
| Sequence | String: FADDLLDLLT FPGAHRFLHK PTRNSSSLYS RANNNFDVGV LPGYPTKNVN LFSPLTNSTL PINGLHRSYQ PLMLNCLTKI TNHTLSMYL LPSEIQTYSC GGAMVKYQTH DAVRIILDLT ATDHISVEVV GQHGENYVFV CSEQFNYTTA LHNSTFFSLN S ELYCFTNN ...String: FADDLLDLLT FPGAHRFLHK PTRNSSSLYS RANNNFDVGV LPGYPTKNVN LFSPLTNSTL PINGLHRSYQ PLMLNCLTKI TNHTLSMYL LPSEIQTYSC GGAMVKYQTH DAVRIILDLT ATDHISVEVV GQHGENYVFV CSEQFNYTTA LHNSTFFSLN S ELYCFTNN TYLGILPPDL TDFTVYRTGQ FYANGYLLGT LPITVNYVRL YRGHLSANSA HFALANLTDT LITLTNTTIS QI TYCDKSV VDSIACQRSS HEVEDGFYSD PKSAVRARQR TIVTLPKLPE LEVVQLNISA HMDFGEARLD SVTINGNTSY CVT KPYFRL ETNFMCTGCT MNLRTDTCSF DLSAVNNGMS FSQFCLSTES GACEMKIIVT YVWNYLLRQR LYVTAVEGQT HTGT TSVHA TDTSSVITDV CTDYTIYGVS GTGIIKPSDL LLHNGIAFTS PTGELYAFKN ITTGKTLQVL PCETPSQLIV INNTV VGAI TSSNSTENNR FTTTIVTPTF FYSTNATTFN CTKPVLSYGP ISVCSDGAIV GISTLQNTRP SIVSLYDGEV EIPSAF SLS VQTEYLQVQA EQVIVDCPQY VCNGNSRCLQ LLAQYTSACS NIEAALHSSA QLDSREIINM FKTSTQSLQL ANITNFK GD YNFSSILTTR IGGRSAIEDL LFNKVVTSGL GTVDQDYKSC SRDMAIADLV CSQYYNGIMV LPGVVDAEKM AMYTGSLT G AMVFGGLTAA AAIPFATAVQ ARLNYVALQT NVLQENQKIL AESFNQAVGN ISLALSSVND AIQQTSEALN TVAIAIKKI QTVVNQQGEA LSHLTAQLSN NFQAISTSIQ DIYNRLEEVE ANQQVDRLIT GRLAALNAYV TQLLNQMSQI RQSRLLAQQK INECVKSQS SRYGFCGNGT HIFSLTQTAP NGIFFMHAVL VPNKFTRVNA SAGICVDNTR GYSLQPQLIL YQFNNSWRVT P RNMYEPRL PRQADFIQLT DCSVTFYNTT AANLPNIIPD IIDVNQTVSD IIDNLPTATP PQWDVGIYNN TILNLTVEIN DL QERSKNL SQIADRLQNY IDNVDIKQIE DKIEEILSKI YHIENEIARI KKLIGEIGGG GSHHHHHHHH UniProtKB: Spike protein |
-Macromolecule #4: 2-acetamido-2-deoxy-beta-D-glucopyranose
| Macromolecule | Name: 2-acetamido-2-deoxy-beta-D-glucopyranose / type: ligand / ID: 4 / Number of copies: 24 / Formula: NAG |
|---|---|
| Molecular weight | Theoretical: 221.208 Da |
| Chemical component information |  ChemComp-NAG: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.35 mg/mL |
|---|---|
| Buffer | pH: 7.2 |
| Grid | Model: C-flat-2/1 / Pretreatment - Type: GLOW DISCHARGE |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Average electron dose: 1.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
