 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3895 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | S.aureus ClpC resting state, asymmetric map | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | ClpC / AAA+ protease / oligomeric complex / CHAPERONE | |||||||||
| Function / homology |  Function and homology information Function and homology informationcellular response to heat / ATP hydrolysis activity / ATP binding / cytoplasm Similarity search - Function | |||||||||
| Biological species |   Staphylococcus aureus (bacteria) / Staphylococcus aureus (strain bovine RF122 / ET3-1) (bacteria) Staphylococcus aureus (bacteria) / Staphylococcus aureus (strain bovine RF122 / ET3-1) (bacteria) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 8.4 Å | |||||||||
 Authors Authors | Carroni M / Mogk A | |||||||||
 Citation Citation | Journal: Elife / Year: 2017 Title: Regulatory coiled-coil domains promote head-to-head assemblies of AAA+ chaperones essential for tunable activity control. Authors: Marta Carroni / Kamila B Franke / Michael Maurer / Jasmin Jäger / Ingo Hantke / Felix Gloge / Daniela Linder / Sebastian Gremer / Kürşad Turgay / Bernd Bukau / Axel Mogk /   Abstract: Ring-forming AAA+ chaperones exert ATP-fueled substrate unfolding by threading through a central pore. This activity is potentially harmful requiring mechanisms for tight repression and substrate- ...Ring-forming AAA+ chaperones exert ATP-fueled substrate unfolding by threading through a central pore. This activity is potentially harmful requiring mechanisms for tight repression and substrate-specific activation. The AAA+ chaperone ClpC with the peptidase ClpP forms a bacterial protease essential to virulence and stress resistance. The adaptor MecA activates ClpC by targeting substrates and stimulating ClpC ATPase activity. We show how ClpC is repressed in its ground state by determining ClpC cryo-EM structures with and without MecA. ClpC forms large two-helical assemblies that associate via head-to-head contacts between coiled-coil middle domains (MDs). MecA converts this resting state to an active planar ring structure by binding to MD interaction sites. Loss of ClpC repression in MD mutants causes constitutive activation and severe cellular toxicity. These findings unravel an unexpected regulatory concept executed by coiled-coil MDs to tightly control AAA+ chaperone activity. | |||||||||
| History |
|
- Structure visualization
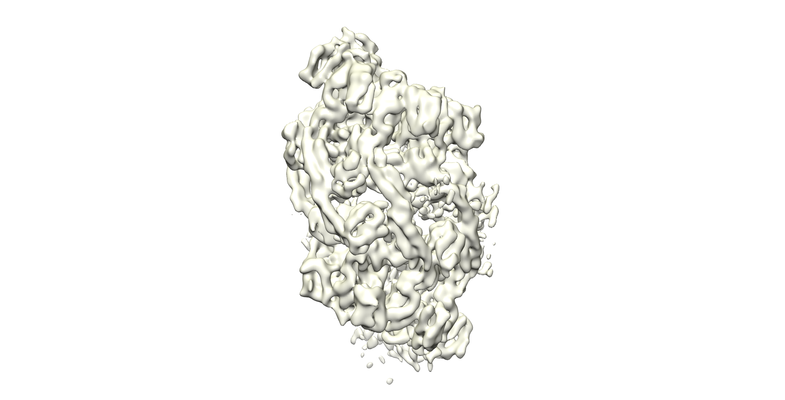
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3895.map.gz | 96.6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3895-v30.xmlemd-3895.xml | 16.7 KB 16.7 KB | Display Display | EMDB header |
| Images | 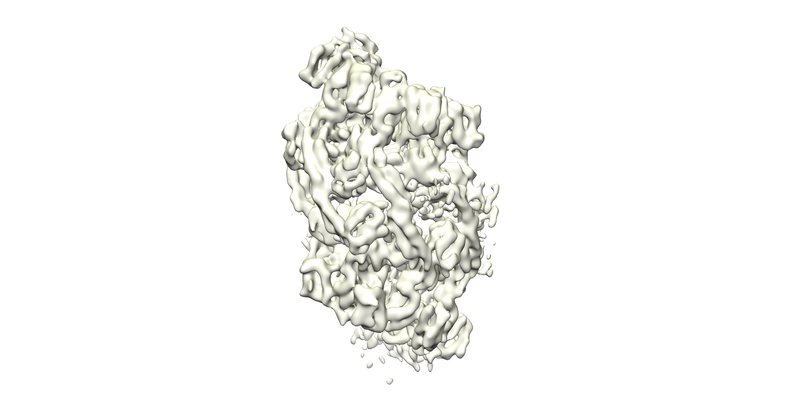 emd_3895.png emd_3895.png | 114.1 KB | ||
| Masks | emd_3895_msk_1.map | 103 MB | Mask map | |
| Filedesc metadata | emd-3895.cif.gz | 6.1 KB | ||
| Others | emd_3895_half_map_1.map.gzemd_3895_half_map_2.map.gz | 95.8 MB 95.8 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3895ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3895 http://ftp.pdbj.org/pub/emdb/structures/EMD-3895ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3895 | HTTPS FTP |
-Related structure data
| Related structure data |  6em9MC  3894C  3897C  6em8C  6emwC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |







-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |



-Map
| File | Download / File: emd_3895.map.gz / Format: CCP4 / Size: 103 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
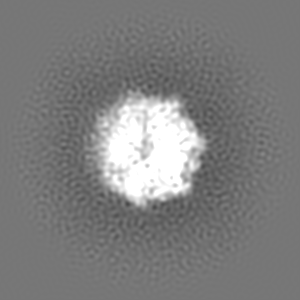
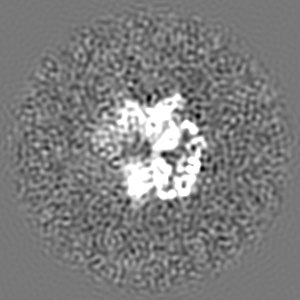
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.34 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-Supplemental data
-Mask #1
| File | emd_3895_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #1
| File | emd_3895_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |
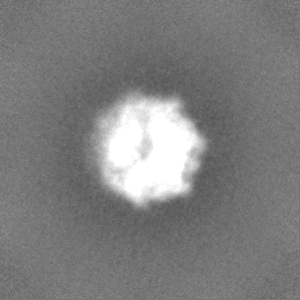
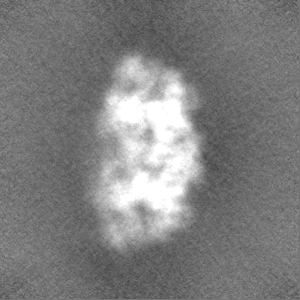

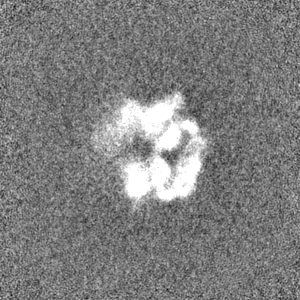
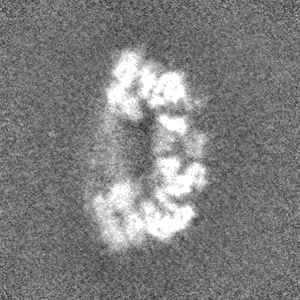

-Half map: #2
| File | emd_3895_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |
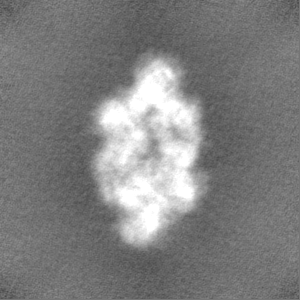
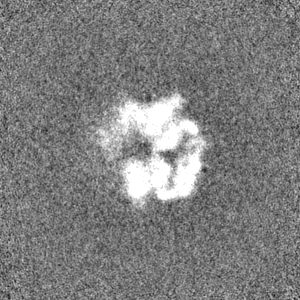
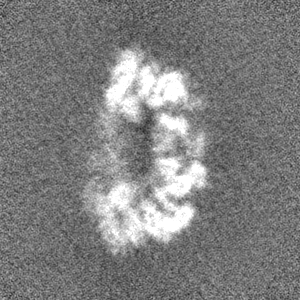
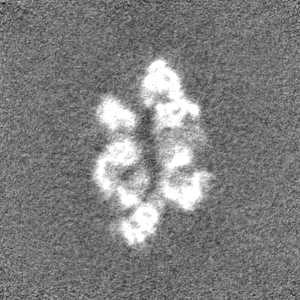
- Sample components
Sample components
-Entire : Resting-state oligomeric complex of S. aureus ClpC
| Entire | Name: Resting-state oligomeric complex of S. aureus ClpC |
|---|---|
| Components |
|
-Supramolecule #1: Resting-state oligomeric complex of S. aureus ClpC
| Supramolecule | Name: Resting-state oligomeric complex of S. aureus ClpC / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Staphylococcus aureus (bacteria) / Strain: bovine RF122 / ET3-1 |
| Molecular weight | Theoretical: 900 KDa |
-Macromolecule #1: ATP-dependent Clp protease ATP-binding subunit ClpC
| Macromolecule | Name: ATP-dependent Clp protease ATP-binding subunit ClpC / type: protein_or_peptide / ID: 1 / Number of copies: 10 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Staphylococcus aureus (strain bovine RF122 / ET3-1) (bacteria) Strain: bovine RF122 / ET3-1 |
| Molecular weight | Theoretical: 91.200375 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MLFGRLTERA QRVLAHAQEE AIRLNHSNIG TEHLLLGLMK EPEGIAAKVL ESFNITEDKV IEEVEKLIGH GQDHVGTLHY TPRAKKVIE LSMDEARKLH HNFVGTEHIL LGLIRENEGV AARVFANLDL NITKARAQVV KALGNPEMSN KNAQASKSNN T PTLDSLAR ...String: MLFGRLTERA QRVLAHAQEE AIRLNHSNIG TEHLLLGLMK EPEGIAAKVL ESFNITEDKV IEEVEKLIGH GQDHVGTLHY TPRAKKVIE LSMDEARKLH HNFVGTEHIL LGLIRENEGV AARVFANLDL NITKARAQVV KALGNPEMSN KNAQASKSNN T PTLDSLAR DLTVIAKDGT LDPVIGRDKE ITRVIEVLSR RTKNNPVLIG EPGVGKTAIA EGLAQAIVNN EVPETLKDKR VM SLDMGTV VAGTKYRGEF EERLKKVMEE IQQAGNVILF IDELHTLVGA GGAEGAIDAS NILKPALARG ELQCIGATTL DEY RKNIEK DAALERRFQP VQVDEPSVVD TVAILKGLRD RYEAHHRINI SDEAIEAAVK LSNRYVSDRF LPDKAIDLID EASS KVRLK SHTTPNNLKE IEQEIEKVKN EKDAAVHAQE FENAANLRDK QTKLEKQYEE AKNEWKNTQN GMSTSLSEED IAEVI AGWT GIPLTKINET ESEKLLSLED TLHERVIGQK DAVNSISKAV RRARAGLKDP KRPIGSFIFL GPTGVGKTEL ARALAE SMF GDDDAMIRVD MSEFMEKHAV SRLVGAPPGY VGHDDGGQLT EKVRRKPYSV ILFDEIEKAH PDVFNILLQV LDDGHLT DT KGRTVDFRNT IIIMTSNVGA QELQDQRFAG FGGSSDGQDY ETIRKTMLKE LKNSFRPEFL NRVDDIIVFH KLTKEELK E IVTMMVNKLT NRLSEQNINI IVTDKAKDKI AEEGYDPEYG ARPLIRAIQK TIEDNLSELI LDGNQIEGKK VTVDHDGKE FKYDIAEQTS ETKTPSQA UniProtKB: ATP-dependent Clp protease ATP-binding subunit ClpC |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.5 |
|---|---|
| Grid | Model: C-flat / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 40 sec. / Pretreatment - Atmosphere: AIR |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 297 K / Instrument: FEI VITROBOT MARK IV |
| Details | concentration used was 6uM |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON II (4k x 4k) / Detector mode: INTEGRATING / Average electron dose: 30.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 3.0 µm / Nominal defocus min: 1.0 µm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Startup model | Type of model: OTHER / Details: Stochastic Gradient Descent |
|---|---|
| Final reconstruction | Applied symmetry - Point group: C1 (asymmetric) / Resolution.type: BY AUTHOR / Resolution: 8.4 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 2.6) / Number images used: 40000 |
| Initial angle assignment | Type: RANDOM ASSIGNMENT |
| Final angle assignment | Type: PROJECTION MATCHING / Software - Name: RELION (ver. 2.3) |
-Atomic model buiding 1
| Details | Phyre2 base on the crystal structure PDB: 3pxi |
|---|---|
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT |
| Output model | PDB-6em9: |
-Atomic model buiding 2
| Details | Phyre2 base on the crystal structure PDB: 3pxi |
|---|---|
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT |
| Output model | PDB-6em9: |
