 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-3453 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Cryo-EM structure of human p53 bound to a molecular support structure made of DNA origami. | |||||||||
 マップデータ マップデータ | None | |||||||||
 試料 試料 |
| |||||||||
| 生物種 |  Homo sapiens (ヒト) Homo sapiens (ヒト) | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 15.0 Å | |||||||||
 データ登録者 データ登録者 | Martin TG / Bharat TAM / Joerger AC / Bai X / Praetorius F / Fersht AR / Dietz H / Scheres SHW | |||||||||
 引用 引用 | ジャーナル: Proc Natl Acad Sci U S A / 年: 2016 タイトル: Design of a molecular support for cryo-EM structure determination. 著者: Thomas G Martin / Tanmay A M Bharat / Andreas C Joerger / Xiao-Chen Bai / Florian Praetorius / Alan R Fersht / Hendrik Dietz / Sjors H W Scheres /   要旨: Despite the recent rapid progress in cryo-electron microscopy (cryo-EM), there still exist ample opportunities for improvement in sample preparation. Macromolecular complexes may disassociate or ...Despite the recent rapid progress in cryo-electron microscopy (cryo-EM), there still exist ample opportunities for improvement in sample preparation. Macromolecular complexes may disassociate or adopt nonrandom orientations against the extended air-water interface that exists for a short time before the sample is frozen. We designed a hollow support structure using 3D DNA origami to protect complexes from the detrimental effects of cryo-EM sample preparation. For a first proof-of-principle, we concentrated on the transcription factor p53, which binds to specific DNA sequences on double-stranded DNA. The support structures spontaneously form monolayers of preoriented particles in a thin film of water, and offer advantages in particle picking and sorting. By controlling the position of the binding sequence on a single helix that spans the hollow support structure, we also sought to control the orientation of individual p53 complexes. Although the latter did not yet yield the desired results, the support structures did provide partial information about the relative orientations of individual p53 complexes. We used this information to calculate a tomographic 3D reconstruction, and refined this structure to a final resolution of ∼15 Å. This structure settles an ongoing debate about the symmetry of the p53 tetramer bound to DNA. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
 ムービービューア ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_3453.map.gz | 1.6 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-3453-v30.xmlemd-3453.xml | 12.5 KB 12.5 KB | 表示 表示 | EMDBヘッダ |
| 画像 | 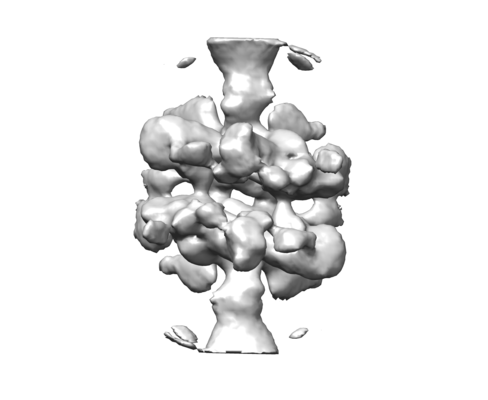 emd_3453.png emd_3453.png | 45.6 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3453ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3453 http://ftp.pdbj.org/pub/emdb/structures/EMD-3453ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3453 | HTTPS FTP |
-関連構造データ






-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_3453.map.gz / 形式: CCP4 / 大きさ: 2.8 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | None | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.76 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-添付データ
- 試料の構成要素
試料の構成要素
-全体 : Tetramer of truncated human p53 (residues 1-360) bound to dsDNA
| 全体 | 名称: Tetramer of truncated human p53 (residues 1-360) bound to dsDNA |
|---|---|
| 要素 |
|
-超分子 #1: Tetramer of truncated human p53 (residues 1-360) bound to dsDNA
| 超分子 | 名称: Tetramer of truncated human p53 (residues 1-360) bound to dsDNA タイプ: complex / ID: 1 / 親要素: 0 詳細: The dsDNA to which p53 was bound is part of a large DNA origami support structure that was used in the structure determination process. |
|---|---|
| 由来(天然) | 生物種: Homo sapiens (ヒト) |
| 組換発現 | 生物種:  |
| 分子量 | 理論値: 160 KDa |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 緩衝液 | pH: 7.5 |
|---|---|
| グリッド | モデル: Quantifoil R2/2 / 材質: COPPER / メッシュ: 200 / 支持フィルム - 材質: CARBON / 支持フィルム - トポロジー: HOLEY ARRAY / 前処理 - タイプ: GLOW DISCHARGE / 前処理 - 雰囲気: AIR |
| 凍結 | 凍結剤: ETHANE |
| 詳細 | P53 tetramers were bound to large DNA origami support structures. |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 特殊光学系 | エネルギーフィルター - 名称: GIF エネルギーフィルター - エネルギー下限: -20 eV エネルギーフィルター - エネルギー上限: +20 eV |
| 撮影 | フィルム・検出器のモデル: GATAN K2 SUMMIT (4k x 4k) 検出モード: SUPER-RESOLUTION / デジタル化 - サイズ - 横: 3710 pixel / デジタル化 - サイズ - 縦: 3710 pixel / デジタル化 - 画像ごとのフレーム数: 1-20 / 撮影したグリッド数: 7 / 実像数: 2562 / 平均露光時間: 16.0 sec. / 平均電子線量: 38.0 e/Å2 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | C2レンズ絞り径: 70.0 µm / 最大 デフォーカス(補正後): 5.0 µm / 最小 デフォーカス(補正後): 1.0 µm / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER ホルダー冷却材: NITROGEN |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
