 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-3026: Electron cryo-microscopy of porcine Factor VIII bound to lipid na... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3026 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Electron cryo-microscopy of porcine Factor VIII bound to lipid nanotubes and single particle tomography reconstruction | |||||||||
 Map data Map data | SPT reconstruction from tomograms of negatively stained porcine FVIII bound to LNT in a helical organization. The volume is low pass filter to 20 Angstroms in EMAN2 | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Blood coagulation factor VIII / Lipid nanotubes / Cryo-electron microscopy / Membrane-bound organization / Single particle tomography reconstruction | |||||||||
| Biological species |  | |||||||||
| Method | subtomogram averaging / negative staining / Resolution: 21.0 Å | |||||||||
 Authors Authors | Dalm D / Galaz-Montoya JG / Miller JL / Grushin K / Villalobos A / Koyfman AY / Schmid MF / Stoilova-McPhie S | |||||||||
 Citation Citation | Journal: J Struct Biol / Year: 2015 Title: Single particle tomography in EMAN2. Authors: Jesús G Galaz-Montoya / John Flanagan / Michael F Schmid / Steven J Ludtke /  Abstract: Single particle tomography (SPT or subtomogram averaging) offers a powerful alternative to traditional 2-D single particle reconstruction for studying conformationally or compositionally ...Single particle tomography (SPT or subtomogram averaging) offers a powerful alternative to traditional 2-D single particle reconstruction for studying conformationally or compositionally heterogeneous macromolecules. It can also provide direct observation (without labeling or staining) of complexes inside cells at nanometer resolution. The development of computational methods and tools for SPT remains an area of active research. Here we present the EMAN2.1 SPT toolbox, which offers a full SPT processing pipeline, from particle picking to post-alignment analysis of subtomogram averages, automating most steps. Different algorithm combinations can be applied at each step, providing versatility and allowing for procedural cross-testing and specimen-specific strategies. Alignment methods include all-vs-all, binary tree, iterative single-model refinement, multiple-model refinement, and self-symmetry alignment. An efficient angular search, Graphic Processing Unit (GPU) acceleration and both threaded and distributed parallelism are provided to speed up processing. Finally, automated simulations, per particle reconstruction of subtiltseries, and per-particle Contrast Transfer Function (CTF) correction have been implemented. Processing examples using both real and simulated data are shown for several structures. | |||||||||
| History |
|
- Structure visualization
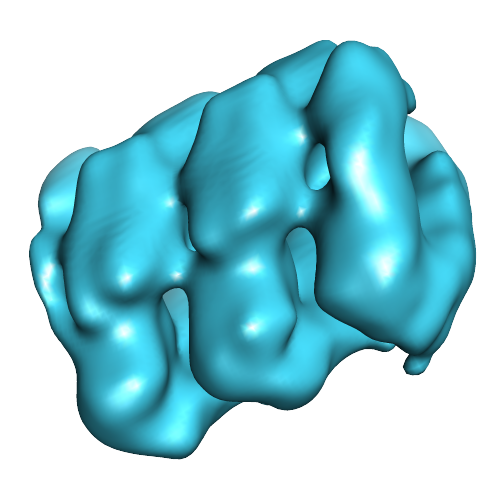
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3026.map.gz | 1.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3026-v30.xmlemd-3026.xml | 12.7 KB 12.7 KB | Display Display | EMDB header |
| Images |  emd_3026.png emd_3026.png | 143.8 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3026ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3026 http://ftp.pdbj.org/pub/emdb/structures/EMD-3026ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3026 | HTTPS FTP |
-Related structure data







-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_3026.map.gz / Format: CCP4 / Size: 3.3 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | SPT reconstruction from tomograms of negatively stained porcine FVIII bound to LNT in a helical organization. The volume is low pass filter to 20 Angstroms in EMAN2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 2.9 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Helically organized porcine blood coagulation Factor VIII bound t...
| Entire | Name: Helically organized porcine blood coagulation Factor VIII bound to a lipid nanotube |
|---|---|
| Components |
|
-Supramolecule #1000: Helically organized porcine blood coagulation Factor VIII bound t...
| Supramolecule | Name: Helically organized porcine blood coagulation Factor VIII bound to a lipid nanotube type: sample / ID: 1000 Details: The protein was monodisperse in solution and bound to the lipid nanotube in 1:1 w/w ratio to achieve helical organization Oligomeric state: dimer / Number unique components: 2 |
|---|---|
| Molecular weight | Experimental: 170 KDa / Theoretical: 170 KDa / Method: SDS gel and amino acid sequence |
-Macromolecule #1: Blood coagulation Factor VIII
| Macromolecule | Name: Blood coagulation Factor VIII / type: protein_or_peptide / ID: 1 / Name.synonym: Hemophilia A factor Details: Recombinant porcine Factor VIII lacking the B domain was bound to lipid nanotubes and helically organized for structure analysis by cryo-EM. Number of copies: 4 / Oligomeric state: 2 / Recombinant expression: Yes |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Experimental: 170 KDa / Theoretical: 170 KDa |
| Recombinant expression | Organism:  Cricetulus griseus (Chinese hamster) / Recombinant cell: BHK Cricetulus griseus (Chinese hamster) / Recombinant cell: BHK |
-Macromolecule #2: LIPID NANOTUBE
| Macromolecule | Name: LIPID NANOTUBE / type: ligand / ID: 2 / Recombinant expression: No / Database: NCBI |
|---|---|
| Source (natural) | Organism: unidentified (others) |
-Experimental details
-Structure determination
| Method | negative staining |
|---|---|
 Processing Processing | subtomogram averaging |
| Aggregation state | helical array |
-Sample preparation
| Concentration | 1.0 mg/mL |
|---|---|
| Buffer | pH: 7.4 / Details: 20 mM HEPES, 150 mM NaCL, 5 mM CaCl2 |
| Staining | Type: NEGATIVE Details: The pFVIII-LNT ehlcial tubes were absorbed on amorphous carbon and stained with 1% Uranyl acetate |
| Grid | Details: Evaporated carbon on mice was floated on 300 mesh copper grids, dried, glow discharged for 10 seconds |
| Vitrification | Cryogen name: NONE / Instrument: OTHER |
| Details | The protein and lipid are mixed in 1:1 ration and incubated for 15 minutes |
- Electron microscopy
Electron microscopy
| Microscope | JEOL 2100 |
|---|---|
| Date | Oct 10, 2014 |
| Image recording | Category: CCD / Film or detector model: GATAN ULTRASCAN 4000 (4k x 4k) / Average electron dose: 20 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source: LAB6 |
| Electron optics | Calibrated magnification: 52000 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.0 mm / Nominal defocus max: -0.0025 µm / Nominal defocus min: -0.001 µm / Nominal magnification: 40000 |
| Sample stage | Specimen holder model: SIDE ENTRY, EUCENTRIC / Tilt series - Axis1 - Min angle: 2 ° / Tilt series - Axis1 - Max angle: 60 ° |
-Image processing
| Details | described in details in the submitted references |
|---|---|
| Final reconstruction | Applied symmetry - Helical parameters - Δz: 36 Å Applied symmetry - Helical parameters - Δ&Phi: 35.5 ° Applied symmetry - Helical parameters - Axial symmetry: C5 (5 fold cyclic) Resolution.type: BY AUTHOR / Resolution: 21.0 Å / Resolution method: OTHER / Software - Name: EMAN2 / Number subtomograms used: 756 |
-Atomic model buiding 1
| Initial model | PDB ID: Chain - #0 - Chain ID: A / Chain - #1 - Chain ID: B |
|---|---|
| Software | Name:  UCSF Chimera UCSF Chimera |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |

