 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-29699: Structure of cobalamin-dependent methionine synthase (MetH) in a ... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of cobalamin-dependent methionine synthase (MetH) in a resting state | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Methyltransferase / Transferase / Amino-acid biosynthesis / Methionine biosynthesis | |||||||||
| Function / homology |  Function and homology information Function and homology informationmethionine synthase / methionine synthase activity / homocysteine metabolic process / tetrahydrofolate metabolic process / cobalamin binding / methylation / zinc ion binding / cytosol Similarity search - Function | |||||||||
| Biological species |   Thermus filiformis (bacteria) Thermus filiformis (bacteria) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.6 Å | |||||||||
 Authors Authors | Watkins MB / Ando N | |||||||||
| Funding support |  United States, 2 items United States, 2 items
| |||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2023 Title: Conformational switching and flexibility in cobalamin-dependent methionine synthase studied by small-angle X-ray scattering and cryoelectron microscopy. Authors: Maxwell B Watkins / Haoyue Wang / Audrey Burnim / Nozomi Ando / Abstract: Cobalamin-dependent methionine synthase (MetH) catalyzes the synthesis of methionine from homocysteine and 5-methyltetrahydrofolate (CH-Hfolate) using the unique chemistry of its cofactor. In doing ...Cobalamin-dependent methionine synthase (MetH) catalyzes the synthesis of methionine from homocysteine and 5-methyltetrahydrofolate (CH-Hfolate) using the unique chemistry of its cofactor. In doing so, MetH links the cycling of -adenosylmethionine with the folate cycle in one-carbon metabolism. Extensive biochemical and structural studies on MetH have shown that this flexible, multidomain enzyme adopts two major conformations to prevent a futile cycle of methionine production and consumption. However, as MetH is highly dynamic as well as both a photosensitive and oxygen-sensitive metalloenzyme, it poses special challenges for structural studies, and existing structures have necessarily come from a "divide and conquer" approach. In this study, we investigate MetH and a thermophilic homolog from using small-angle X-ray scattering (SAXS), single-particle cryoelectron microscopy (cryo-EM), and extensive analysis of the AlphaFold2 database to present a structural description of the full-length MetH in its entirety. Using SAXS, we describe a common resting-state conformation shared by both active and inactive oxidation states of MetH and the roles of CH-Hfolate and flavodoxin in initiating turnover and reactivation. By combining SAXS with a 3.6-Å cryo-EM structure of the MetH, we show that the resting-state conformation consists of a stable arrangement of the catalytic domains that is linked to a highly mobile reactivation domain. Finally, by combining AlphaFold2-guided sequence analysis and our experimental findings, we propose a general model for functional switching in MetH. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_29699.map.gz | 203.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-29699-v30.xmlemd-29699.xml | 21.5 KB 21.5 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_29699_fsc.xml | 12.9 KB | Display | FSC data file |
| Images | 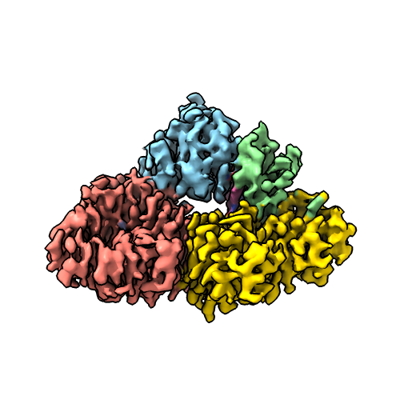 emd_29699.png emd_29699.png | 100.4 KB | ||
| Masks | emd_29699_msk_1.map | 216 MB | Mask map | |
| Filedesc metadata | emd-29699.cif.gz | 7.6 KB | ||
| Others | emd_29699_half_map_1.map.gzemd_29699_half_map_2.map.gz | 200.7 MB 200.7 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-29699ftp://ftp.pdbj.org/pub/emdb/structures/EMD-29699 http://ftp.pdbj.org/pub/emdb/structures/EMD-29699ftp://ftp.pdbj.org/pub/emdb/structures/EMD-29699 | HTTPS FTP |
-Related structure data
| Related structure data |  8g3hMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |
-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_29699.map.gz / Format: CCP4 / Size: 216 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.0691 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Mask #1
| File | emd_29699_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #2
| File | emd_29699_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #1
| File | emd_29699_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








- Sample components
Sample components
-Entire : Cobalamin-dependent methionine synthase holoprotein
| Entire | Name: Cobalamin-dependent methionine synthase holoprotein |
|---|---|
| Components |
|
-Supramolecule #1: Cobalamin-dependent methionine synthase holoprotein
| Supramolecule | Name: Cobalamin-dependent methionine synthase holoprotein / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 Details: Cobalamin-dependent methionine synthase (MetH) in complex with B12 and Zinc |
|---|---|
| Source (natural) | Organism: Thermus filiformis (bacteria) |
| Molecular weight | Theoretical: 132.539 KDa |
-Macromolecule #1: Methionine synthase
| Macromolecule | Name: Methionine synthase / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO / EC number: methionine synthase |
|---|---|
| Source (natural) | Organism: Thermus filiformis (bacteria) |
| Molecular weight | Theoretical: 131.312266 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MVEVHTCSPG CRHHLGGAGW GDAPLLRLGY NKEARARRFP YLRALSQRVL VFDGAMGTEL QKRDLTPEDY GEEAYYGCPE VLNRTRPDV VREIHLSYLE AGAEVIETNS FGALRHVLAE YGLGEEAEEL AYRAARIAKE AAEPYGAFVA GALGPGTKLV S LGQISWEE ...String: MVEVHTCSPG CRHHLGGAGW GDAPLLRLGY NKEARARRFP YLRALSQRVL VFDGAMGTEL QKRDLTPEDY GEEAYYGCPE VLNRTRPDV VREIHLSYLE AGAEVIETNS FGALRHVLAE YGLGEEAEEL AYRAARIAKE AAEPYGAFVA GALGPGTKLV S LGQISWEE LFSAYKEAVR GLVRGGVDLI LLETAQDILQ VRCAVLAARE AMAELGVELP LQVQVTFEAT GTMLVGTDEQ AA LAALESL PIDVVGMNCA TGPDLMDAKI RYFAQESTRF VACLPNAGLP RNEGGRVVYD LTPEELARWH LKFVTEYGVN AVG GCCGTG PEHIRKVAEA VGGRPSPVHK TAFPPQVASL YQAVPLRQET SLLLVGERLN ATGSKRFREL LFAGDLEGIL ALAQ EQVAE GAHVLDLSVA WTGRDELEDM KRVLSKLATG VTVPFMVDST SPEVMEEALK RLPGRAILNS ANLEDGLEKF DRVAS LAKA HGAALVALAI DEEGMAKTRV KKVEVALRMY ERLTEHHGLR PEDLLFDLLT FPITQGDEET RPLARETLLA LEELRE RLP GVGFVLGVSN VSFGLKPKAR RVLNSVFLDE ARKKGLTAAI VDAGKILPIS QIPEEAYALA LDLIYDRREG QDPLFAF IR FFEEHKEVLA EDKEAFQALP VEERLRRRVL EGKRVGLEED LAEALGRMRP LEIINGPLLE AMKEVGELFG AGKMQLPF V LQAAEVMKQA VAYLEPHMEK KGEGKGKLVL ATVKGDVHDI GKNLVDIILT NNGYTVYNLG IKVPIEEMLK AVDEVKPHA LGMSGLLVKS TQVMKENLEY MRARGYTLPV ILGGAALTRA YVEELRSIYP EVYYAEDAFE GLALMEELTG HRPKDLTLRR ARTTRKEAP APRSKPVSPA PDLPRPPFFG VRVEEGLDLA TIAHYVNRLA LYRGQWGFSR GGLSREEWEA YVKREADPVF R RLLAEAMA EGWLRPRVLY GFFPVAREGE ELWVYSPEGE LLEKLRFPRQ RGGGISLVDY FRPRFAEPLS DEADWLPGYE EG ARDVLGV QLVTMGEEPK RKAEALYREG RYQDYLFVHG FAVEMTEALA EYWHKRMRQM WGIAGQDAPE IRRLFQQEYR GAR YSFGYP ACPDLADQAK LDRLMDFSRI GVRLTENYQL DPEHSTSALV VHHPEARYFS VD UniProtKB: Methionine synthase |
-Macromolecule #2: ZINC ION
| Macromolecule | Name: ZINC ION / type: ligand / ID: 2 / Number of copies: 1 / Formula: ZN |
|---|---|
| Molecular weight | Theoretical: 65.409 Da |
-Macromolecule #3: COBALAMIN
| Macromolecule | Name: COBALAMIN / type: ligand / ID: 3 / Number of copies: 1 / Formula: B12 |
|---|---|
| Molecular weight | Theoretical: 1.330356 KDa |
| Chemical component information | 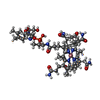 ChemComp-B12: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | 2D array |
-Sample preparation
| Concentration | 0.264 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.6 Component:
Details: 50 mM HEPES, 150 mM NaCl, 2.5 mM DTT pH 7.6 | ||||||||||||
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: CONTINUOUS / Pretreatment - Type: GLOW DISCHARGE | ||||||||||||
| Vitrification | Cryogen name: ETHANE-PROPANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV | ||||||||||||
| Details | Sample was mixed with equimolar commercial horse spleen apoferritin for freezing. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Bioquantum / Energy filter - Slit width: 30 eV |
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Digitization - Dimensions - Width: 5760 pixel / Digitization - Dimensions - Height: 5092 pixel / Number grids imaged: 2 / Number real images: 12768 / Average exposure time: 2.5 sec. / Average electron dose: 65.0 e/Å2 Details: Images were collected in movie mode with a total of 50 frames over 2.5 seconds. |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 0.8 µm / Nominal magnification: 81000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Initial model | PDB ID: Chain - Chain ID: A / Chain - Residue range: 2-896 / Chain - Source name: AlphaFold / Chain - Initial model type: in silico model |
|---|---|
| Details | Initial model used was the 3 N-terminal domains of AlphaFold database model A0A1Q9SZ17. Each domain was fit individually by rigid-body fitting in Chimera. The cobalamin ligand was initially fit by alignment of the 1BMT crystal structure to the appropriate binding region. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |
| Output model | PDB-8g3h: |

