 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | MicroED structure of proteinase K recorded on K2 | |||||||||
 マップデータ マップデータ | 2mFo-DFc | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード | serine protease / HYDROLASE | |||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報phosphorelay signal transduction system / regulation of DNA-templated transcription / DNA binding / membrane 類似検索 - 分子機能 | |||||||||
| 生物種 |  Parengyodontium album (菌類) Parengyodontium album (菌類) | |||||||||
| 手法 | 電子線結晶学 / クライオ電子顕微鏡法 | |||||||||
 データ登録者 データ登録者 | Clabbers MTB / Martynowycz MW / Hattne J / Nannenga BL / Gonen T | |||||||||
| 資金援助 |  米国, 2件 米国, 2件
| |||||||||
 引用 引用 | ジャーナル: J Struct Biol / 年: 2022 タイトル: Electron-counting MicroED data with the K2 and K3 direct electron detectors. 著者: Max T B Clabbers / Michael W Martynowycz / Johan Hattne / Brent L Nannenga / Tamir Gonen / 要旨: Microcrystal electron diffraction (MicroED) uses electron cryo-microscopy (cryo-EM) to collect diffraction data from small crystals during continuous rotation of the sample. As a result of advances ...Microcrystal electron diffraction (MicroED) uses electron cryo-microscopy (cryo-EM) to collect diffraction data from small crystals during continuous rotation of the sample. As a result of advances in hardware as well as methods development, the data quality has continuously improved over the past decade, to the point where even macromolecular structures can be determined ab initio. Detectors suitable for electron diffraction should ideally have fast readout to record data in movie mode, and high sensitivity at low exposure rates to accurately report the intensities. Direct electron detectors are commonly used in cryo-EM imaging for their sensitivity and speed, but despite their availability are generally not used in diffraction. Primary concerns with diffraction experiments are the dynamic range and coincidence loss, which will corrupt the measurement if the flux exceeds the count rate of the detector. Here, we describe instrument setup and low-exposure MicroED data collection in electron-counting mode using K2 and K3 direct electron detectors and show that the integrated intensities can be effectively used to solve structures of two macromolecules between 1.2 Å and 2.8 Å resolution. Even though a beam stop was not used with the K3 studies we did not observe damage to the camera. As these cameras are already available in many cryo-EM facilities, this provides opportunities for users who do not have access to dedicated facilities for MicroED. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 添付画像 |
|---|
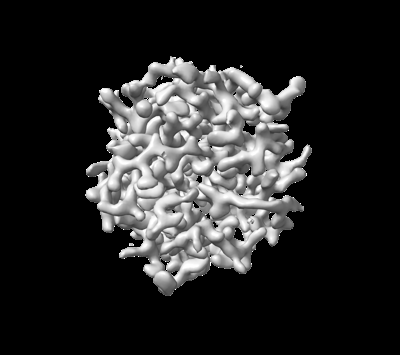
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_27900.map.gz | 2 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-27900-v30.xmlemd-27900.xml | 13.5 KB 13.5 KB | 表示 表示 | EMDBヘッダ |
| 画像 |  emd_27900.png emd_27900.png | 52.9 KB | ||
| Filedesc metadata | emd-27900.cif.gz | 5.5 KB | ||
| Filedesc structureFactors | emd_27900_sf.cif.gz | 167.1 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-27900ftp://ftp.pdbj.org/pub/emdb/structures/EMD-27900 http://ftp.pdbj.org/pub/emdb/structures/EMD-27900ftp://ftp.pdbj.org/pub/emdb/structures/EMD-27900 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_27900_validation.pdf.gz | 578 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_27900_full_validation.pdf.gz | 577.5 KB | 表示 | |
| XML形式データ | emd_27900_validation.xml.gz | 4.4 KB | 表示 | |
| CIF形式データ | emd_27900_validation.cif.gz | 5 KB | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-27900ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-27900 | HTTPS FTP |
-関連構造データ




-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |

-マップ
| ファイル | ダウンロード / ファイル: emd_27900.map.gz / 形式: CCP4 / 大きさ: 2.2 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | 2mFo-DFc | ||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 これらの図は立方格子座標系で作成されたものです | ||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X: 0.6757 Å / Y: 0.6757 Å / Z: 0.6307 Å | ||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
|
 X (Sec.)
X (Sec.) Y (Row.)
Y (Row.) Z (Col.)
Z (Col.)





















-添付データ
- 試料の構成要素
試料の構成要素
-全体 : Proteinase K
| 全体 | 名称: Proteinase K |
|---|---|
| 要素 |
|
-超分子 #1: Proteinase K
| 超分子 | 名称: Proteinase K / タイプ: complex / ID: 1 / 親要素: 0 / 含まれる分子: #1 / 詳細: Serine protease |
|---|---|
| 由来(天然) | 生物種: Parengyodontium album (菌類) |
| 分子量 | 理論値: 28.9 KDa |
-分子 #1: Proteinase K
| 分子 | 名称: Proteinase K / タイプ: protein_or_peptide / ID: 1 / コピー数: 1 / 光学異性体: LEVO / EC番号: peptidase K |
|---|---|
| 由来(天然) | 生物種: Parengyodontium album (菌類) |
| 分子量 | 理論値: 28.930783 KDa |
| 組換発現 | 生物種: Parengyodontium album (菌類) |
| 配列 | 文字列: AAQTNAPWGL ARISSTSPGT STYYYDESAG QGSCVYVIDT GIEASHPEFE GRAQMVKTYY YSSRDGNGHG THCAGTVGSR TYGVAKKTQ LFGVKVLDDN GSGQYSTIIA GMDFVASDKN NRNCPKGVVA SLSLGGGYSS SVNSAAARLQ SSGVMVAVAA G NNNADARN ...文字列: AAQTNAPWGL ARISSTSPGT STYYYDESAG QGSCVYVIDT GIEASHPEFE GRAQMVKTYY YSSRDGNGHG THCAGTVGSR TYGVAKKTQ LFGVKVLDDN GSGQYSTIIA GMDFVASDKN NRNCPKGVVA SLSLGGGYSS SVNSAAARLQ SSGVMVAVAA G NNNADARN YSPASEPSVC TVGASDRYDR RSSFSNYGSV LDIFGPGTSI LSTWIGGSTR SISGTSMATP HVAGLAAYLM TL GKTTAAS ACRYIADTAN KGDLSNIPFG TVNLLAYNNY QA UniProtKB: Putative transcriptional regulator ToxR |
-分子 #2: CALCIUM ION
| 分子 | 名称: CALCIUM ION / タイプ: ligand / ID: 2 / コピー数: 4 / 式: CA |
|---|---|
| 分子量 | 理論値: 40.078 Da |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 電子線結晶学 |
| 試料の集合状態 | 3D array |
-試料調製
| 濃度 | 50 mg/mL |
|---|---|
| 緩衝液 | pH: 8 |
| グリッド | モデル: Quantifoil R2/1 / 材質: COPPER / メッシュ: 300 / 支持フィルム - 材質: CARBON / 支持フィルム - トポロジー: HOLEY / 支持フィルム - Film thickness: 10 |
| 凍結 | 凍結剤: ETHANE / チャンバー内湿度: 95 % / チャンバー内温度: 277 K / 装置: LEICA PLUNGER |
| 詳細 | Microcrystals |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 温度 | 最低: 77.0 K / 最高: 90.0 K |
| 撮影 | フィルム・検出器のモデル: GATAN K2 BASE (4k x 4k) デジタル化 - サイズ - 横: 1650 pixel / デジタル化 - サイズ - 縦: 1470 pixel / 撮影したグリッド数: 1 / 実像数: 1 / 回折像の数: 1600 / 平均露光時間: 0.025 sec. / 平均電子線量: 0.01 e/Å2 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | C2レンズ絞り径: 50.0 µm / 照射モード: FLOOD BEAM / 撮影モード: DIFFRACTION / Cs: 2.7 mm / カメラ長: 590 mm |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER ホルダー冷却材: NITROGEN |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
-画像解析
| 最終 再構成 | 解像度の算出法: DIFFRACTION PATTERN/LAYERLINES / ソフトウェア - 名称: REFMAC |
|---|---|
| Merging software list | ソフトウェア - 名称: AIMLESS |
| Crystallography statistics | Number intensities measured: 30326 / Number structure factors: 5453 / Fourier space coverage: 83 / R sym: 30 / R merge: 64 / Overall phase error: 28 / Overall phase residual: 0 / Phase error rejection criteria: None / High resolution: 2.7 Å / 殻 - Shell ID: 1 / 殻 - High resolution: 2.7 Å / 殻 - Low resolution: 3.4 Å / 殻 - Number structure factors: 2499 / 殻 - Phase residual: 33 / 殻 - Fourier space coverage: 83 / 殻 - Multiplicity: 6 |
-原子モデル構築 1
| 精密化 | 空間: RECIPROCAL / プロトコル: RIGID BODY FIT / 温度因子: 20.32 / 当てはまり具合の基準: Maximum liklihood |
|---|---|
| 得られたモデル |  PDB-8e52: |
