 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-2435 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
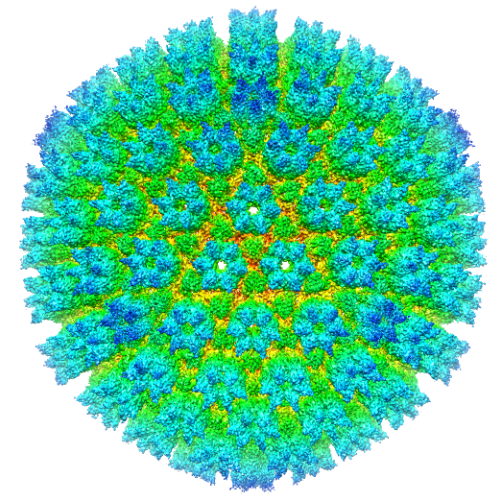
| タイトル | Protein Interactions in the Murine Cytomegalovirus Capsid Revealed by CryoEM | |||||||||
 マップデータ マップデータ | Reconstruction of Murine Cytomegalovirus | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード | cytomegalovirus / herpes simplex virus type 1 / electron cryo microscopy / three-dimensional / major capsid protein. | |||||||||
| 生物種 |  Murid herpesvirus 1 (ヘルペスウイルス) Murid herpesvirus 1 (ヘルペスウイルス) | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / ネガティブ染色法 / 解像度: 9.1 Å | |||||||||
 データ登録者 データ登録者 | Hui W / Tang Q / Liu H / Atanasov I / Zhu H / Zhou ZH | |||||||||
 引用 引用 | ジャーナル: Protein Cell / 年: 2013 タイトル: Protein interactions in the murine cytomegalovirus capsid revealed by cryoEM. 著者: Wong H Hui / Qiyi Tang / Hongrong Liu / Ivo Atanasov / Fenyong Liu / Hua Zhu / Z Hong Zhou /  要旨: Cytomegalovirus (CMV) is distinct among members of the Herpesviridae family for having the largest dsDNA genome (230 kb). Packaging of large dsDNA genome is known to give rise to a highly pressurized ...Cytomegalovirus (CMV) is distinct among members of the Herpesviridae family for having the largest dsDNA genome (230 kb). Packaging of large dsDNA genome is known to give rise to a highly pressurized viral capsid, but molecular interactions conducive to the formation of CMV capsid resistant to pressurization have not been described. Here, we report a cryo electron microscopy (cryoEM) structure of the murine cytomegalovirus (MCMV) capsid at a 9.1 Å resolution and describe the molecular interactions among the ∼3000 protein molecules in the MCMV capsid at the secondary structure level. Secondary structural elements are resolved to provide landmarks for correlating with results from sequence-based prediction and for structure-based homology modeling. The major capsid protein (MCP) upper domain (MCPud) contains α-helices and β-sheets conserved with those in MCPud of herpes simplex virus type 1 (HSV-1), with the largest differences identified as a "saddle loop" region, located at the tip of MCPud and involved in interaction with the smallest capsid protein (SCP). Interactions among the bacteriophage HK97-like floor domain of MCP, the middle domain of MCP, the hook and clamp domains of the triplex proteins (hoop and clamp domains of TRI-1 and clamp domain of TRI-2) contribute to the formation of a mature capsid. These results offer a framework for understanding how cytomegalovirus uses various secondary structural elements of its capsid proteins to build a robust capsid for packaging its large dsDNA genome inside and for attaching unique functional tegument proteins outside. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
 ムービービューア ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_2435.map.gz | 754.6 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-2435-v30.xmlemd-2435.xml | 13.2 KB 13.2 KB | 表示 表示 | EMDBヘッダ |
| 画像 |  emd_2435.png emd_2435.png | 495.6 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-2435ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2435 http://ftp.pdbj.org/pub/emdb/structures/EMD-2435ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2435 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_2435_validation.pdf.gz | 236.1 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_2435_full_validation.pdf.gz | 235.2 KB | 表示 | |
| XML形式データ | emd_2435_validation.xml.gz | 4.7 KB | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-2435ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-2435 | HTTPS FTP |
-関連構造データ
| 類似構造データ |
|---|

-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_2435.map.gz / 形式: CCP4 / 大きさ: 817.7 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Reconstruction of Murine Cytomegalovirus | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 これらの図は立方格子座標系で作成されたものです | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.8 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
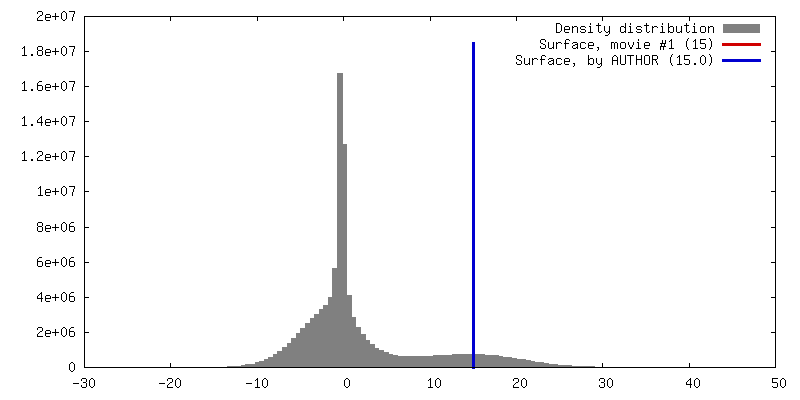
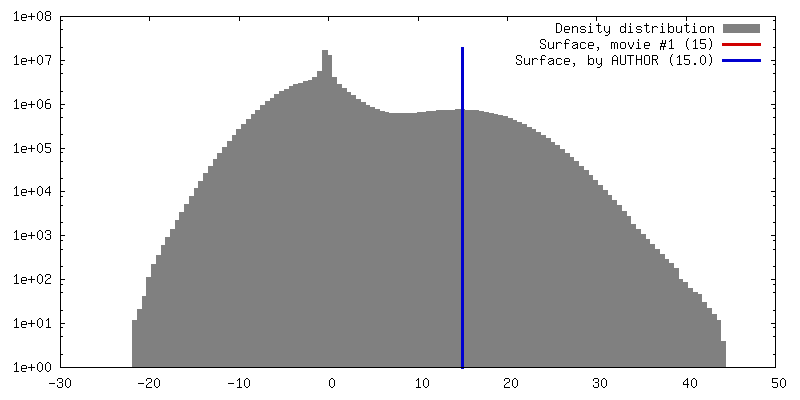
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-添付データ
- 試料の構成要素
試料の構成要素
-全体 : Murine cytomegalovirus capsid
| 全体 | 名称: Murine cytomegalovirus capsid |
|---|---|
| 要素 |
|
-超分子 #1000: Murine cytomegalovirus capsid
| 超分子 | 名称: Murine cytomegalovirus capsid / タイプ: sample / ID: 1000 / 詳細: The sample is monodisperse in PBS buffer / 集合状態: icosahedral virus capsid / Number unique components: 4 |
|---|---|
| 分子量 | 理論値: 186.4 MDa |
-超分子 #1: Murid herpesvirus 1
| 超分子 | 名称: Murid herpesvirus 1 / タイプ: virus / ID: 1 / Name.synonym: Murine cytomegalovirus 詳細: The virus is enveloped but the structure presented here is for the capsid only. NCBI-ID: 10366 / 生物種: Murid herpesvirus 1 / Sci species strain: strain Smith / ウイルスタイプ: OTHER / ウイルス・単離状態: STRAIN / ウイルス・エンベロープ: Yes / ウイルス・中空状態: Yes / Syn species name: Murine cytomegalovirus |
|---|---|
| 宿主 | 生物種: Murine / 株: strain Smith / 別称: INVERTEBRATES |
| 分子量 | 理論値: 186.4 MDa |
| ウイルス殻 | Shell ID: 1 / 名称: Capsid / 直径: 1310 Å / T番号(三角分割数): 16 |
-実験情報
-構造解析
| 手法 | ネガティブ染色法, クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 1 mg/mL |
|---|---|
| 緩衝液 | pH: 7 / 詳細: PBS |
| 染色 | タイプ: NEGATIVE / 詳細: vitreous ice, no staining |
| グリッド | 詳細: across holes in Quantifoil grids |
| 凍結 | 凍結剤: ETHANE / チャンバー内湿度: 100 % / チャンバー内温度: 90 K / 装置: FEI VITROBOT MARK I / 詳細: Vitrification instrument: FEI Vitrobot / 手法: Blot for 7-9 seconds before plunging |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 温度 | 最低: 80 K / 最高: 100 K / 平均: 90 K |
| アライメント法 | Legacy - 非点収差: objective lens astigmatism was corrected at 250,000 times magnification Legacy - Electron beam tilt params: 0 |
| 特殊光学系 | エネルギーフィルター - 名称: FEI |
| 日付 | 2009年10月15日 |
| 撮影 | カテゴリ: FILM / フィルム・検出器のモデル: KODAK SO-163 FILM デジタル化 - スキャナー: NIKON SUPER COOLSCAN 9000 デジタル化 - サンプリング間隔: 6.35 µm / 実像数: 1299 / 平均電子線量: 20 e/Å2 / ビット/ピクセル: 16 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2.7 mm / 最大 デフォーカス(公称値): 3.0 µm / 最小 デフォーカス(公称値): 1.5 µm / 倍率(公称値): 47000 |
| 試料ステージ | 試料ホルダー: Autoloader of Titan Krios at liquid nitrogen temperature 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
-画像解析
| 詳細 | Individual particle images were automatically boxed out from micrographs by the autoBox program in the IMIRS package, followed by manual screening to select good capsid particle images that appear perfectly intact and without contamination and signs of specimen charging. Defocus value and astigmatism parameters of each micrographs were determined with CTFFIND. Subsequent data processing includes the determination of particle orientation/center parameters, 3D reconstruction and iterative, projection-based refinement by a distributed computing approach using modular programs in the IMIRS package with recent enhancements. The distributed computing was performed entirely through on six Microsoft Windows personal computers and four Windows servers within a custom-designed MPI network. Astigmatism was taken into consideration during the correction of contrast transfer function (CTF) both in the orientation/center refinement step and the 3D reconstruction step. Using this procedure, we first obtained a 3D map from 6402 particle images from the Polara micrographs. This map has a resolution of about 11 angstrom and was used as the starting model to assist processing the higher resolution particle images of the Titan micrographs. To process the Titan micrographs, we discarded micrographs with specimen charging by evaluating the Fourier transform of the micrographs and selected 669 micrographs for in-depth data processing. From the 669 good Titan micrographs selected out through this process, we boxed 5383 particle images and determined their orientation/center parameters by using the 11 angstrom map as the starting model. These orientation/center parameters were iteratively refined against the latest 3D map by gradually including Fourier data at regions of higher spatial frequency. The iterative process was terminated when the reconstruction converges to a stable solution and no further improvement in the resolution of the map was observed. Our reconstruction converges when the high spatial frequency cut-off of the included image data reached 1/6.9 angstrom-1. The final map was obtained from 3467 particles images, all from the Titan micrographs. |
|---|---|
| CTF補正 | 詳細: CTFFIND |
| 最終 再構成 | 想定した対称性 - 点群: I (正20面体型対称) / アルゴリズム: OTHER / 解像度のタイプ: BY AUTHOR / 解像度: 9.1 Å / 解像度の算出法: FSC 0.5 CUT-OFF / ソフトウェア - 名称: IMIRS / 使用した粒子像数: 3467 |
-原子モデル構築 1
| 初期モデル | PDB ID: Chain - Chain ID: A |
|---|---|
| ソフトウェア | 名称: Chimera |
| 詳細 | Manual fitting in Chimera |
| 精密化 | 空間: REAL / プロトコル: RIGID BODY FIT / 当てはまり具合の基準: Chimera fit-to-map |

