ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-23772 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
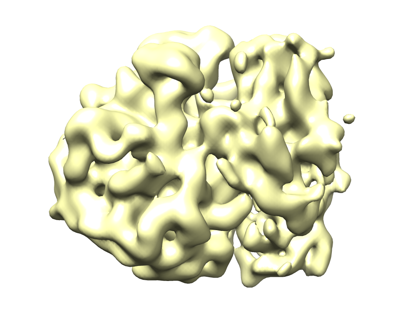
| タイトル | 3D reconstruction generated using the locations and orientations of 1,172 50S subunits detected in 220 images using 2DTM with a B. subtilis 50S template | |||||||||
 マップデータ マップデータ | 20-Angstrom filtered reconstruction | |||||||||
 試料 試料 |
| |||||||||
| 生物種 |  Mycoplasma pneumoniae (バクテリア) Mycoplasma pneumoniae (バクテリア) | |||||||||
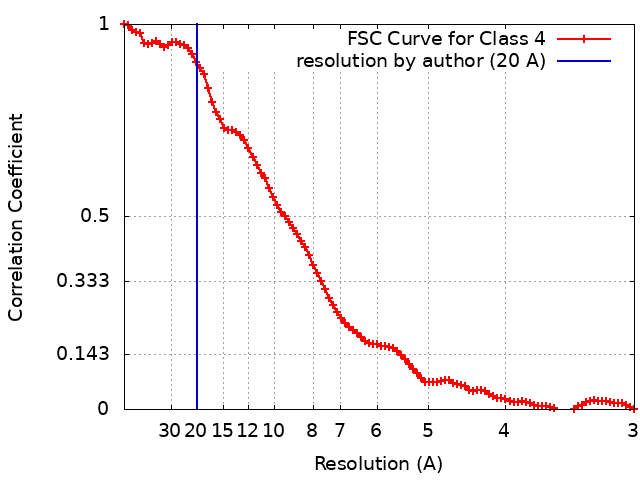
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 20.0 Å | |||||||||
 データ登録者 データ登録者 | Lucas BA / Himes BA / Xue L / Grant T / Mahamid J / Grigorieff N | |||||||||
| 資金援助 |  ドイツ, 1件 ドイツ, 1件
| |||||||||
 引用 引用 | ジャーナル: Elife / 年: 2021 タイトル: Locating macromolecular assemblies in cells by 2D template matching with cisTEM. 著者: Bronwyn A Lucas / Benjamin A Himes / Liang Xue / Timothy Grant / Julia Mahamid / Nikolaus Grigorieff /  要旨: For a more complete understanding of molecular mechanisms, it is important to study macromolecules and their assemblies in the broader context of the cell. This context can be visualized at nanometer ...For a more complete understanding of molecular mechanisms, it is important to study macromolecules and their assemblies in the broader context of the cell. This context can be visualized at nanometer resolution in three dimensions (3D) using electron cryo-tomography, which requires tilt series to be recorded and computationally aligned, currently limiting throughput. Additionally, the high-resolution signal preserved in the raw tomograms is currently limited by a number of technical difficulties, leading to an increased false-positive detection rate when using 3D template matching to find molecular complexes in tomograms. We have recently described a 2D template matching approach that addresses these issues by including high-resolution signal preserved in single-tilt images. A current limitation of this approach is the high computational cost that limits throughput. We describe here a GPU-accelerated implementation of 2D template matching in the image processing software TEM that allows for easy scaling and improves the accessibility of this approach. We apply 2D template matching to identify ribosomes in images of frozen-hydrated cells with high precision and sensitivity, demonstrating that this is a versatile tool for in situ visual proteomics and in situ structure determination. We benchmark the results with 3D template matching of tomograms acquired on identical sample locations and identify strengths and weaknesses of both techniques, which offer complementary information about target localization and identity. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
 ムービービューア ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_23772.map.gz | 59.1 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-23772-v30.xmlemd-23772.xml | 19.3 KB 19.3 KB | 表示 表示 | EMDBヘッダ |
| FSC (解像度算出) | emd_23772_fsc.xml | 9.9 KB | 表示 | FSCデータファイル |
| 画像 |  emd_23772.png emd_23772.png | 89.1 KB | ||
| その他 | emd_23772_additional_1.map.gzemd_23772_half_map_1.map.gzemd_23772_half_map_2.map.gz | 59.3 MB 16.7 MB 16.7 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-23772ftp://ftp.pdbj.org/pub/emdb/structures/EMD-23772 http://ftp.pdbj.org/pub/emdb/structures/EMD-23772ftp://ftp.pdbj.org/pub/emdb/structures/EMD-23772 | HTTPS FTP |
-関連構造データ
| 関連構造データ | C: 同じ文献を引用 ( |
|---|---|
| 類似構造データ | |
| 電子顕微鏡画像生データ | EMPIAR-10727 (タイトル: Locating Macromolecular Assemblies in Cells by 2D Template Matching with cisTEM Data size: 11.0 Data #1: Exposure-weighted micrographs of M. pneumoniae [micrographs - single frame] Data #2: Particle stack and cisTEM star file for 50S ribosomal subunits identified in the associated 220 images using the M. pneumoniae 50S template [picked particles - single frame - unprocessed] Data #3: Particle stack and cisTEM star file for 50S ribosomal subunits identified in the associated 220 images using the B. subtilis 50S template [picked particles - single frame - processed]) |










-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |

-マップ
| ファイル | ダウンロード / ファイル: emd_23772.map.gz / 形式: CCP4 / 大きさ: 64 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | 20-Angstrom filtered reconstruction | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.5 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-添付データ
-追加マップ: Unfiltered reconstruction
| ファイル | emd_23772_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Unfiltered reconstruction | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
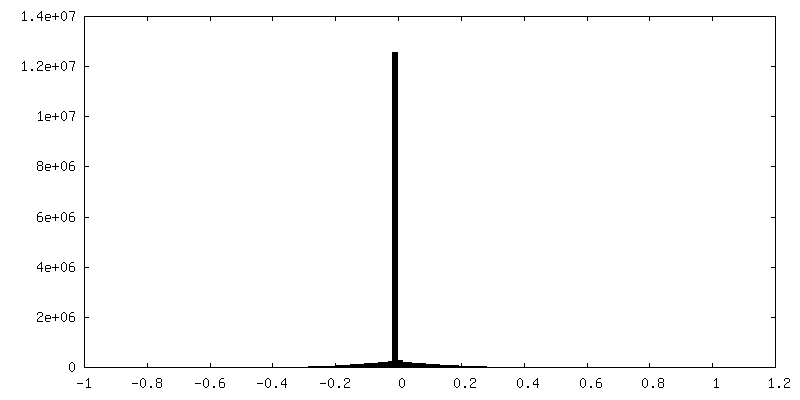
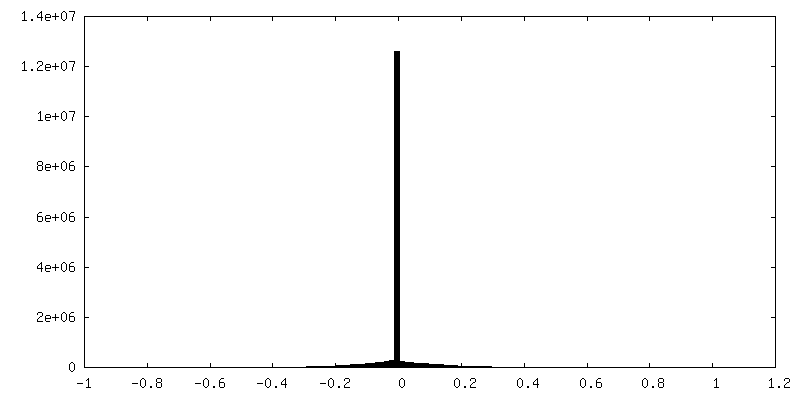
| 密度ヒストグラム |
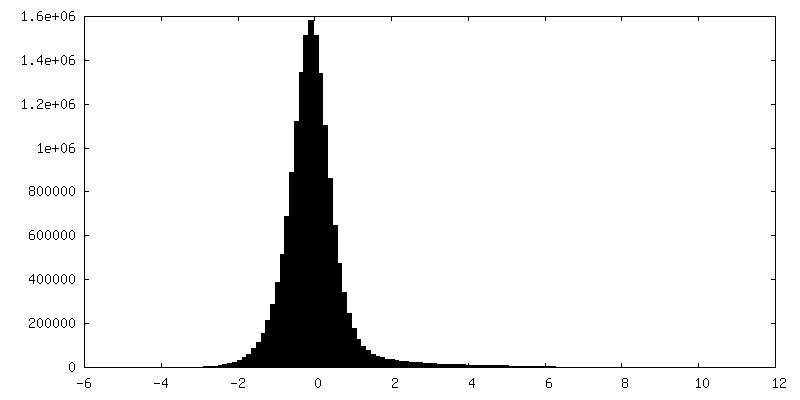
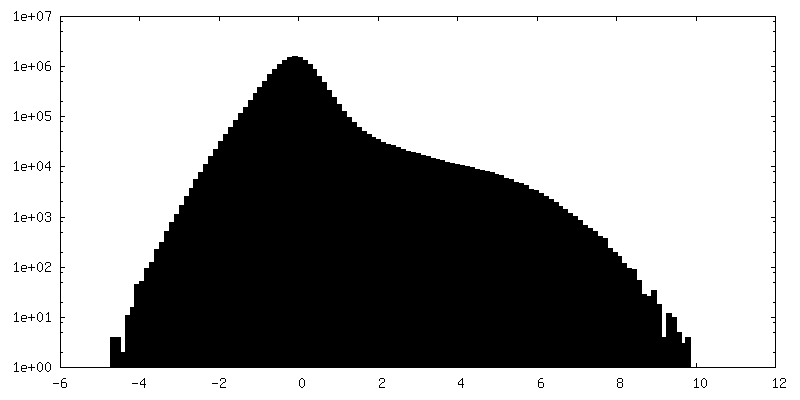
-ハーフマップ: Half map 1
| ファイル | emd_23772_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Half map 1 | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
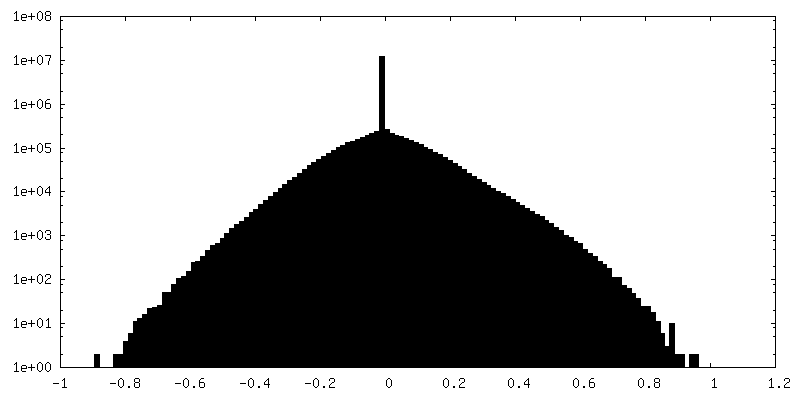
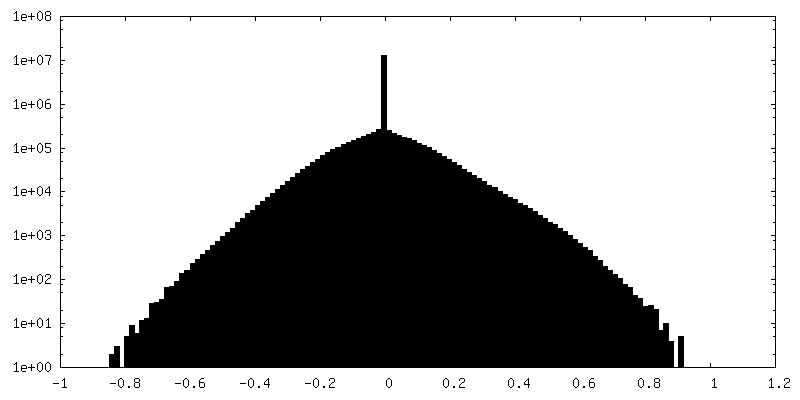
| 密度ヒストグラム |
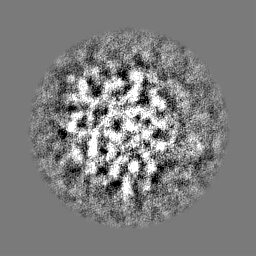
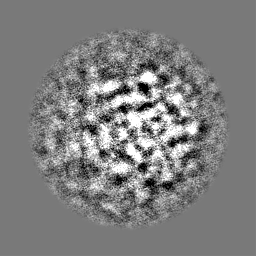
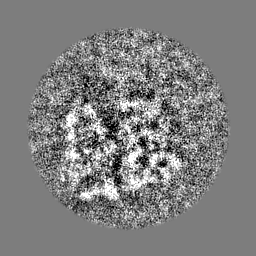
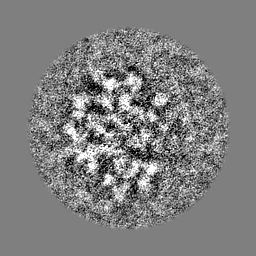
-ハーフマップ: Half map 2
| ファイル | emd_23772_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Half map 2 | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |
- 試料の構成要素
試料の構成要素
-全体 : Mycoplasma pneumoniae 70S ribosome
| 全体 | 名称: Mycoplasma pneumoniae 70S ribosome |
|---|---|
| 要素 |
|
-超分子 #1: Mycoplasma pneumoniae 70S ribosome
| 超分子 | 名称: Mycoplasma pneumoniae 70S ribosome / タイプ: complex / ID: 1 / 親要素: 0 詳細: 3D reconstruction generated using the locations and orientations of 1,172 50S subunits detected in 220 images using 2DTM with a B. subtilis 50S template |
|---|---|
| 由来(天然) | 生物種: Mycoplasma pneumoniae (バクテリア) |
| 分子量 | 理論値: 2.5 MDa |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | cell |
-試料調製
| 緩衝液 | pH: 7 |
|---|---|
| グリッド | モデル: Quantifoil / 材質: GOLD / メッシュ: 200 / 支持フィルム - 材質: CARBON / 支持フィルム - トポロジー: HOLEY ARRAY |
| 凍結 | 凍結剤: ETHANE-PROPANE / チャンバー内湿度: 100 % / チャンバー内温度: 298 K / 装置: FEI VITROBOT MARK IV 詳細: Grids were washed with PBS buffer containing 10 nm protein A-conjugated gold beads (Aurion), blotted from the back side for 2 s and plunged into mixed liquid ethane/propane at liquid N2 ...詳細: Grids were washed with PBS buffer containing 10 nm protein A-conjugated gold beads (Aurion), blotted from the back side for 2 s and plunged into mixed liquid ethane/propane at liquid N2 temperature with a manual plunger.. |
| 詳細 | Whole Mycoplasma pneumoniae cells deposited on a cryo-EM grid |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | TFS KRIOS |
|---|---|
| 特殊光学系 | エネルギーフィルター - 名称: GIF Bioquantum / エネルギーフィルター - スリット幅: 10 eV |
| 撮影 | フィルム・検出器のモデル: GATAN K2 SUMMIT (4k x 4k) 検出モード: SUPER-RESOLUTION / デジタル化 - サイズ - 横: 5760 pixel / デジタル化 - サイズ - 縦: 4092 pixel / デジタル化 - サンプリング間隔: 5.0 µm / デジタル化 - 画像ごとのフレーム数: 1-20 / 実像数: 220 / 平均露光時間: 2.0 sec. / 平均電子線量: 32.0 e/Å2 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2.7 mm / 最大 デフォーカス(公称値): 2.2 µm / 最小 デフォーカス(公称値): 0.5 µm / 倍率(公称値): 215000 |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER ホルダー冷却材: NITROGEN |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |