 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-23771: 3D reconstruction generated using the locations and orientations ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-23771 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | 3D reconstruction generated using the locations and orientations of 5,080 50S subunits detected in 220 images using 2DTM with a M. pneumoniae 50S template | |||||||||
 Map data Map data | 20-Angstrom filtered reconstruction | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  Mycoplasma pneumoniae (Filterable agent of primary atypical pneumonia) Mycoplasma pneumoniae (Filterable agent of primary atypical pneumonia) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 20.0 Å | |||||||||
 Authors Authors | Lucas BA / Himes BA / Xue L / Grant T / Mahamid J / Grigorieff N | |||||||||
| Funding support |  Germany, 1 items Germany, 1 items
| |||||||||
 Citation Citation | Journal: Elife / Year: 2021 Title: Locating macromolecular assemblies in cells by 2D template matching with cisTEM. Authors: Bronwyn A Lucas / Benjamin A Himes / Liang Xue / Timothy Grant / Julia Mahamid / Nikolaus Grigorieff /  Abstract: For a more complete understanding of molecular mechanisms, it is important to study macromolecules and their assemblies in the broader context of the cell. This context can be visualized at nanometer ...For a more complete understanding of molecular mechanisms, it is important to study macromolecules and their assemblies in the broader context of the cell. This context can be visualized at nanometer resolution in three dimensions (3D) using electron cryo-tomography, which requires tilt series to be recorded and computationally aligned, currently limiting throughput. Additionally, the high-resolution signal preserved in the raw tomograms is currently limited by a number of technical difficulties, leading to an increased false-positive detection rate when using 3D template matching to find molecular complexes in tomograms. We have recently described a 2D template matching approach that addresses these issues by including high-resolution signal preserved in single-tilt images. A current limitation of this approach is the high computational cost that limits throughput. We describe here a GPU-accelerated implementation of 2D template matching in the image processing software TEM that allows for easy scaling and improves the accessibility of this approach. We apply 2D template matching to identify ribosomes in images of frozen-hydrated cells with high precision and sensitivity, demonstrating that this is a versatile tool for in situ visual proteomics and in situ structure determination. We benchmark the results with 3D template matching of tomograms acquired on identical sample locations and identify strengths and weaknesses of both techniques, which offer complementary information about target localization and identity. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_23771.map.gz | 58.8 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-23771-v30.xmlemd-23771.xml | 19.3 KB 19.3 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_23771_fsc.xml | 9.9 KB | Display | FSC data file |
| Images | 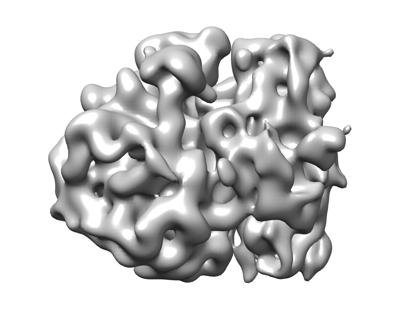 emd_23771.png emd_23771.png | 66.2 KB | ||
| Others | emd_23771_additional_1.map.gzemd_23771_half_map_1.map.gzemd_23771_half_map_2.map.gz | 59.3 MB 16.7 MB 16.6 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-23771ftp://ftp.pdbj.org/pub/emdb/structures/EMD-23771 http://ftp.pdbj.org/pub/emdb/structures/EMD-23771ftp://ftp.pdbj.org/pub/emdb/structures/EMD-23771 | HTTPS FTP |
-Related structure data
| Related structure data | C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10727 (Title: Locating Macromolecular Assemblies in Cells by 2D Template Matching with cisTEM Data size: 11.0 Data #1: Exposure-weighted micrographs of M. pneumoniae [micrographs - single frame] Data #2: Particle stack and cisTEM star file for 50S ribosomal subunits identified in the associated 220 images using the M. pneumoniae 50S template [picked particles - single frame - unprocessed] Data #3: Particle stack and cisTEM star file for 50S ribosomal subunits identified in the associated 220 images using the B. subtilis 50S template [picked particles - single frame - processed]) |










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_23771.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | 20-Angstrom filtered reconstruction | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.5 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Additional map: Unfiltered reconstruction
| File | emd_23771_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unfiltered reconstruction | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |
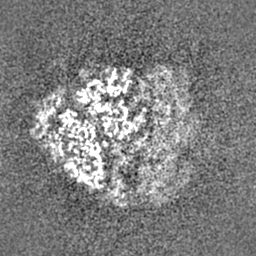
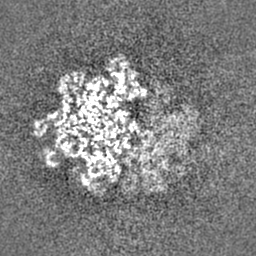
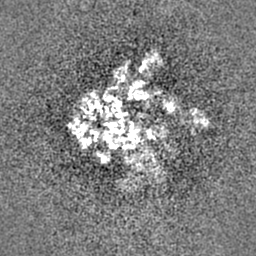
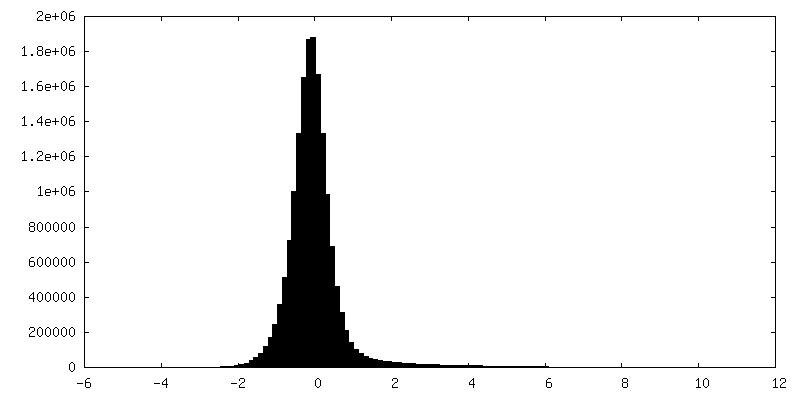
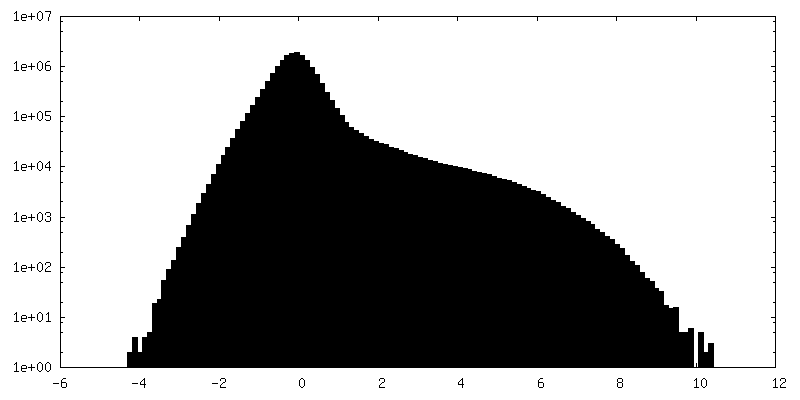
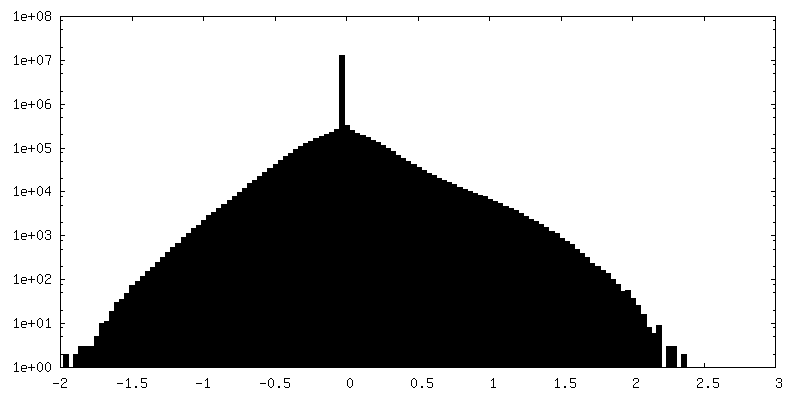
-Half map: Half map 1
| File | emd_23771_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |
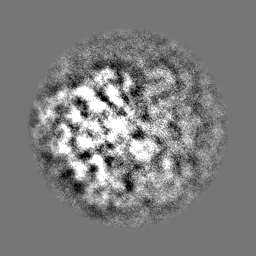
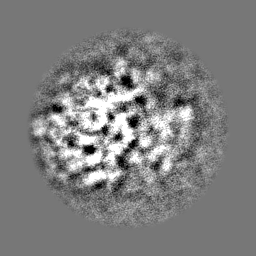
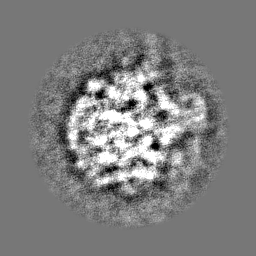


-Half map: Half map 2
| File | emd_23771_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |

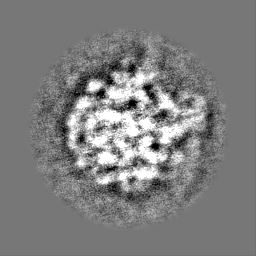

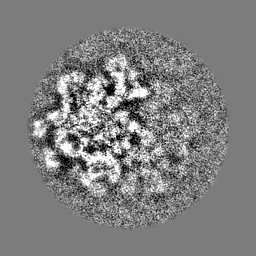
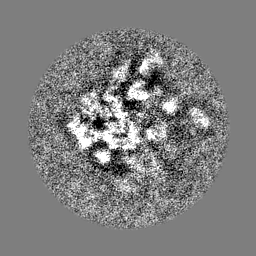
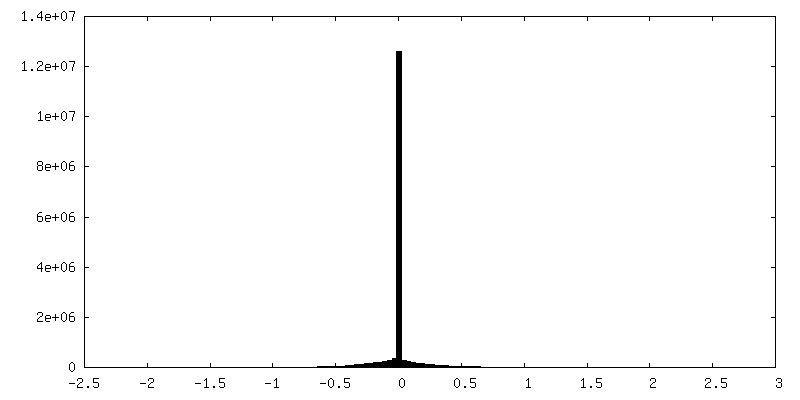

- Sample components
Sample components
-Entire : Mycoplasma pneumoniae 70S ribosome
| Entire | Name: Mycoplasma pneumoniae 70S ribosome |
|---|---|
| Components |
|
-Supramolecule #1: Mycoplasma pneumoniae 70S ribosome
| Supramolecule | Name: Mycoplasma pneumoniae 70S ribosome / type: complex / ID: 1 / Parent: 0 Details: 3D reconstruction generated using the locations and orientations of 5,080 50S subunits detected in 220 images using 2DTM with a M. pneumoniae 50S template |
|---|---|
| Source (natural) | Organism: Mycoplasma pneumoniae (Filterable agent of primary atypical pneumonia) |
| Molecular weight | Theoretical: 2.5 MDa |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | cell |
-Sample preparation
| Buffer | pH: 7 |
|---|---|
| Grid | Model: Quantifoil / Material: GOLD / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY |
| Vitrification | Cryogen name: ETHANE-PROPANE / Chamber humidity: 100 % / Chamber temperature: 298 K / Instrument: FEI VITROBOT MARK IV Details: Grids were washed with PBS buffer containing 10 nm protein A-conjugated gold beads (Aurion), blotted from the back side for 2 s and plunged into mixed liquid ethane/propane at liquid N2 ...Details: Grids were washed with PBS buffer containing 10 nm protein A-conjugated gold beads (Aurion), blotted from the back side for 2 s and plunged into mixed liquid ethane/propane at liquid N2 temperature with a manual plunger.. |
| Details | Whole Mycoplasma pneumoniae cells deposited on a cryo-EM grid |
- Electron microscopy
Electron microscopy
| Microscope | TFS KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Bioquantum / Energy filter - Slit width: 10 eV |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Digitization - Dimensions - Width: 5760 pixel / Digitization - Dimensions - Height: 4092 pixel / Digitization - Sampling interval: 5.0 µm / Digitization - Frames/image: 1-20 / Number real images: 220 / Average exposure time: 2.0 sec. / Average electron dose: 32.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.2 µm / Nominal defocus min: 0.5 µm / Nominal magnification: 215000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |

