National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
R01GM120553
米国
引用
ジャーナル: Proc Natl Acad Sci U S A / 年: 2021 タイトル: Generation of ordered protein assemblies using rigid three-body fusion. 著者: Ivan Vulovic / Qing Yao / Young-Jun Park / Alexis Courbet / Andrew Norris / Florian Busch / Aniruddha Sahasrabuddhe / Hannes Merten / Danny D Sahtoe / George Ueda / Jorge A Fallas / Sara J ...著者: Ivan Vulovic / Qing Yao / Young-Jun Park / Alexis Courbet / Andrew Norris / Florian Busch / Aniruddha Sahasrabuddhe / Hannes Merten / Danny D Sahtoe / George Ueda / Jorge A Fallas / Sara J Weaver / Yang Hsia / Robert A Langan / Andreas Plückthun / Vicki H Wysocki / David Veesler / Grant J Jensen / David Baker / 要旨: Protein nanomaterial design is an emerging discipline with applications in medicine and beyond. A long-standing design approach uses genetic fusion to join protein homo-oligomer subunits via α- ...Protein nanomaterial design is an emerging discipline with applications in medicine and beyond. A long-standing design approach uses genetic fusion to join protein homo-oligomer subunits via α-helical linkers to form more complex symmetric assemblies, but this method is hampered by linker flexibility and a dearth of geometric solutions. Here, we describe a general computational method for rigidly fusing homo-oligomer and spacer building blocks to generate user-defined architectures that generates far more geometric solutions than previous approaches. The fusion junctions are then optimized using Rosetta to minimize flexibility. We apply this method to design and test 92 dihedral symmetric protein assemblies using a set of designed homodimers and repeat protein building blocks. Experimental validation by native mass spectrometry, small-angle X-ray scattering, and negative-stain single-particle electron microscopy confirms the assembly states for 11 designs. Most of these assemblies are constructed from designed ankyrin repeat proteins (DARPins), held in place on one end by α-helical fusion and on the other by a designed homodimer interface, and we explored their use for cryogenic electron microscopy (cryo-EM) structure determination by incorporating DARPin variants selected to bind targets of interest. Although the target resolution was limited by preferred orientation effects and small scaffold size, we found that the dual anchoring strategy reduced the flexibility of the target-DARPIN complex with respect to the overall assembly, suggesting that multipoint anchoring of binding domains could contribute to cryo-EM structure determination of small proteins.
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 マップデータ
マップデータ 試料
試料 データ登録者
データ登録者 米国, 1件
米国, 1件  引用
引用
 構造の表示
構造の表示 ムービービューア
ムービービューア UCSF Chimera
UCSF Chimera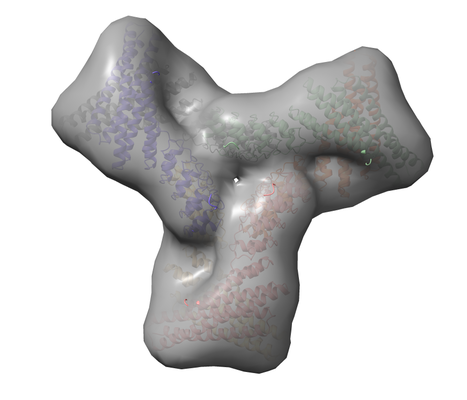
 ダウンロードとリンク
ダウンロードとリンク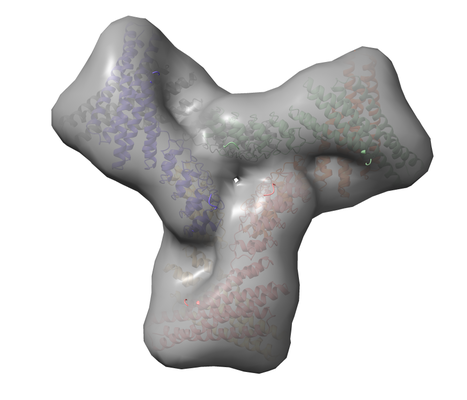 emd_23531.png
emd_23531.png http://ftp.pdbj.org/pub/emdb/structures/EMD-23531
http://ftp.pdbj.org/pub/emdb/structures/EMD-23531
















 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)








































 試料の構成要素
試料の構成要素
 解析
解析 電子顕微鏡法
電子顕微鏡法