 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-21875: ClpP and ClpX IGF loop in ClpX-ClpP complex bound to ssrA tagged GFP -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-21875 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | ClpP and ClpX IGF loop in ClpX-ClpP complex bound to ssrA tagged GFP | |||||||||
 Map data Map data | Phenix sharpened map. | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Protein degradation / AAA+ protease complex / CHAPERONE | |||||||||
| Function / homology |  Function and homology information Function and homology informationprotein denaturation / HslUV protease complex / endopeptidase Clp / endopeptidase Clp complex / positive regulation of programmed cell death / response to temperature stimulus / ATP-dependent peptidase activity / protein quality control for misfolded or incompletely synthesized proteins / protein unfolding / proteasomal protein catabolic process ...protein denaturation / HslUV protease complex / endopeptidase Clp / endopeptidase Clp complex / positive regulation of programmed cell death / response to temperature stimulus / ATP-dependent peptidase activity / protein quality control for misfolded or incompletely synthesized proteins / protein unfolding / proteasomal protein catabolic process / serine-type peptidase activity / proteolysis involved in protein catabolic process / ATP-dependent protein folding chaperone / response to radiation / disordered domain specific binding / unfolded protein binding / ATPase binding / response to heat / protease binding / protein dimerization activity / cell division / serine-type endopeptidase activity / ATP hydrolysis activity / proteolysis / zinc ion binding / ATP binding / identical protein binding / membrane / cytosol Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.88 Å | |||||||||
 Authors Authors | Fei X / Sauer RT | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: Elife / Year: 2020 Title: Structural basis of ClpXP recognition and unfolding of ssrA-tagged substrates. Authors: Xue Fei / Tristan A Bell / Sarah R Barkow / Tania A Baker / Robert T Sauer / Abstract: When ribosomes fail to complete normal translation, all cells have mechanisms to ensure degradation of the resulting partial proteins to safeguard proteome integrity. In and other eubacteria, the ...When ribosomes fail to complete normal translation, all cells have mechanisms to ensure degradation of the resulting partial proteins to safeguard proteome integrity. In and other eubacteria, the tmRNA system rescues stalled ribosomes and adds an ssrA tag or degron to the C-terminus of the incomplete protein, which directs degradation by the AAA+ ClpXP protease. Here, we present cryo-EM structures of ClpXP bound to the ssrA degron. C-terminal residues of the ssrA degron initially bind in the top of an otherwise closed ClpX axial channel and subsequently move deeper into an open channel. For short-degron protein substrates, we show that unfolding can occur directly from the initial closed-channel complex. For longer degron substrates, our studies illuminate how ClpXP transitions from specific recognition into a nonspecific unfolding and translocation machine. Many AAA+ proteases and protein-remodeling motors are likely to employ similar multistep recognition and engagement strategies. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_21875.map.gz | 129.6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-21875-v30.xmlemd-21875.xml | 15.3 KB 15.3 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_21875_fsc.xml | 14.2 KB | Display | FSC data file |
| Images |  emd_21875.png emd_21875.png | 25.4 KB | ||
| Filedesc metadata | emd-21875.cif.gz | 6 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-21875ftp://ftp.pdbj.org/pub/emdb/structures/EMD-21875 http://ftp.pdbj.org/pub/emdb/structures/EMD-21875ftp://ftp.pdbj.org/pub/emdb/structures/EMD-21875 | HTTPS FTP |
-Validation report
| Summary document | emd_21875_validation.pdf.gz | 414.7 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_21875_full_validation.pdf.gz | 414.2 KB | Display | |
| Data in XML | emd_21875_validation.xml.gz | 14.3 KB | Display | |
| Data in CIF | emd_21875_validation.cif.gz | 19.1 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-21875ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-21875 | HTTPS FTP |
-Related structure data
| Related structure data |  6wr2MC  6wrfC  6wsgC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |



-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_21875.map.gz / Format: CCP4 / Size: 244.1 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Phenix sharpened map. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
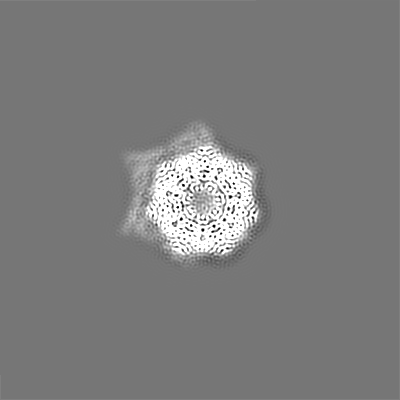
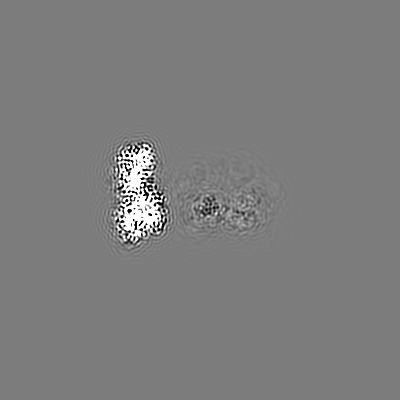
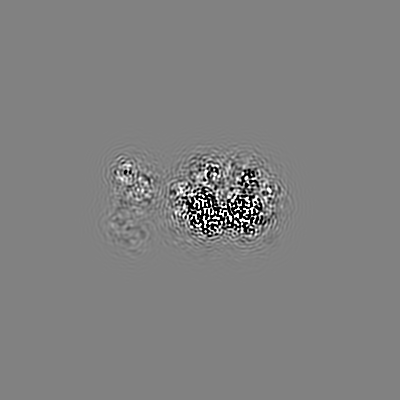
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.87 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
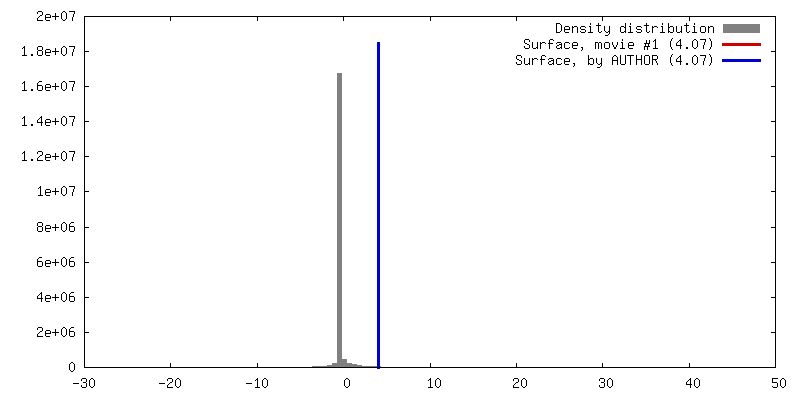
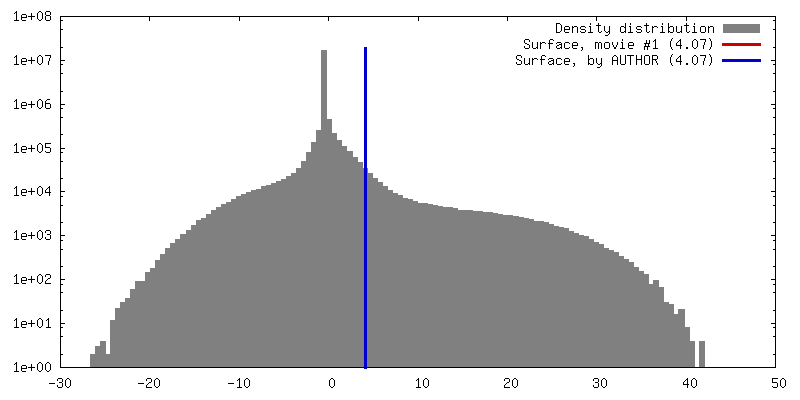
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 X (Sec.)
X (Sec.) Y (Row.)
Y (Row.) Z (Col.)
Z (Col.)















-Supplemental data
- Sample components
Sample components
-Entire : ClpX-ClpP-GFPssrA-ATP/ATPrS
| Entire | Name: ClpX-ClpP-GFPssrA-ATP/ATPrS |
|---|---|
| Components |
|
-Supramolecule #1: ClpX-ClpP-GFPssrA-ATP/ATPrS
| Supramolecule | Name: ClpX-ClpP-GFPssrA-ATP/ATPrS / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#2 |
|---|---|
| Source (natural) | Organism: |
-Macromolecule #1: ATP-dependent Clp protease ATP-binding subunit ClpX
| Macromolecule | Name: ATP-dependent Clp protease ATP-binding subunit ClpX / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 42.355812 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MGSSHHHHHH DYDIPTTENL YFQGSSALPT PHEIRNHLDD YVIGQEQAKK VLAVAVYNHY KRLRNGDTSN GVELGKSNIL LIGPTGSGK TLLAETLARL LDVPFTMADA TTLTEAGYVG EDVENIIQKL LQKSDYDVQK AQRGIVYIDE IDKISRKSDN P SITRDVSG ...String: MGSSHHHHHH DYDIPTTENL YFQGSSALPT PHEIRNHLDD YVIGQEQAKK VLAVAVYNHY KRLRNGDTSN GVELGKSNIL LIGPTGSGK TLLAETLARL LDVPFTMADA TTLTEAGYVG EDVENIIQKL LQKSDYDVQK AQRGIVYIDE IDKISRKSDN P SITRDVSG EGVQQALLKL IEGTVAAVPP QGGRKHPQQE FLQVDTSKIL FICGGAFAGL DKVISHRVET GSGIGFGATV KA KSDKASE GELLAQVEPE DLIKFGLIPE FIGRLPVVAT LNELSEEALI QILKEPKNAL TKQYQALFNL EGVDLEFRDE ALD AIAKKA MARKTGARGL RSIVEAALLD TMYDLPSMED VEKVVIDESV IDGQSEPLLI YGKPEAQQAS GE UniProtKB: ATP-dependent Clp protease ATP-binding subunit ClpX |
-Macromolecule #2: ATP-dependent Clp protease proteolytic subunit
| Macromolecule | Name: ATP-dependent Clp protease proteolytic subunit / type: protein_or_peptide / ID: 2 / Number of copies: 14 / Enantiomer: LEVO / EC number: endopeptidase Clp |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 23.468869 KDa |
| Recombinant expression | Organism: |
| Sequence | String: LVPMVIEQTS RGERSFDIYS RLLKERVIFL TGQVEDHMAN LIVAQMLFLE AENPEKDIYL YINSPGGVIT AGMSIYDTMQ FIKPDVSTI CMGQAASMGA FLLTAGAKGK RFCLPNSRVM IHQPLGGYQG QATDIEIHAR EILKVKGRMN ELMALHTGQS L EQIERDTE ...String: LVPMVIEQTS RGERSFDIYS RLLKERVIFL TGQVEDHMAN LIVAQMLFLE AENPEKDIYL YINSPGGVIT AGMSIYDTMQ FIKPDVSTI CMGQAASMGA FLLTAGAKGK RFCLPNSRVM IHQPLGGYQG QATDIEIHAR EILKVKGRMN ELMALHTGQS L EQIERDTE RDRFLSAPEA VEYGLVDSIL THRNENLYFQ SLEHHHHHH UniProtKB: ATP-dependent Clp protease proteolytic subunit |
-Macromolecule #3: water
| Macromolecule | Name: water / type: ligand / ID: 3 / Number of copies: 14 / Formula: HOH |
|---|---|
| Molecular weight | Theoretical: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.5 Component:
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 60 sec. | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 298 K / Instrument: FEI VITROBOT MARK IV | ||||||||||||
| Details | rapid mixing of ClpX, ClpP, ATPrS with GFPssrA substrate and ATP. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TALOS ARCTICA |
|---|---|
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Detector mode: SUPER-RESOLUTION / Number grids imaged: 1 / Number real images: 4525 / Average exposure time: 60.0 sec. / Average electron dose: 54.0 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: OTHER / Imaging mode: BRIGHT FIELD / Nominal defocus max: -2.5 µm / Nominal defocus min: -0.8 µm / Nominal magnification: 45000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Talos Arctica / Image courtesy: FEI Company |


