Journal: Acta Crystallogr D Struct Biol / Year: 2019 Title: Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Authors: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty ...Authors: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty / Robert D Oeffner / Billy K Poon / Michael G Prisant / Randy J Read / Jane S Richardson / David C Richardson / Massimo D Sammito / Oleg V Sobolev / Duncan H Stockwell / Thomas C Terwilliger / Alexandre G Urzhumtsev / Lizbeth L Videau / Christopher J Williams / Paul D Adams / Abstract: Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological ...Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological processes and to develop new therapeutics against diseases. The overall structure-solution workflow is similar for these techniques, but nuances exist because the properties of the reduced experimental data are different. Software tools for structure determination should therefore be tailored for each method. Phenix is a comprehensive software package for macromolecular structure determination that handles data from any of these techniques. Tasks performed with Phenix include data-quality assessment, map improvement, model building, the validation/rebuilding/refinement cycle and deposition. Each tool caters to the type of experimental data. The design of Phenix emphasizes the automation of procedures, where possible, to minimize repetitive and time-consuming manual tasks, while default parameters are chosen to encourage best practice. A graphical user interface provides access to many command-line features of Phenix and streamlines the transition between programs, project tracking and re-running of previous tasks.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information
 Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) Authors
Authors Poland, 1 items
Poland, 1 items  Citation
Citation


 Structure visualization
Structure visualization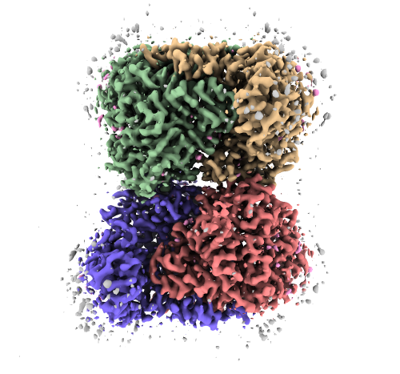
 Downloads & links
Downloads & links emd_18436.png
emd_18436.png http://ftp.pdbj.org/pub/emdb/structures/EMD-18436
http://ftp.pdbj.org/pub/emdb/structures/EMD-18436


 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















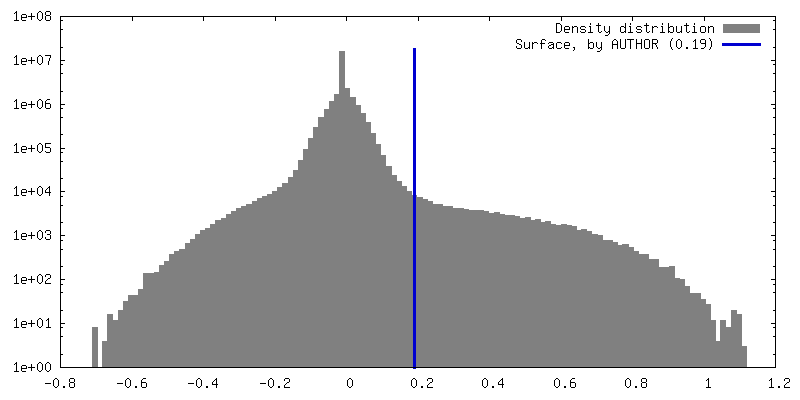
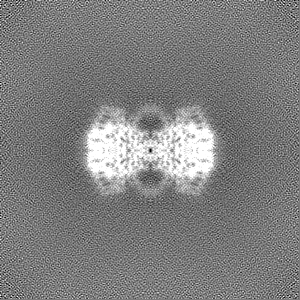





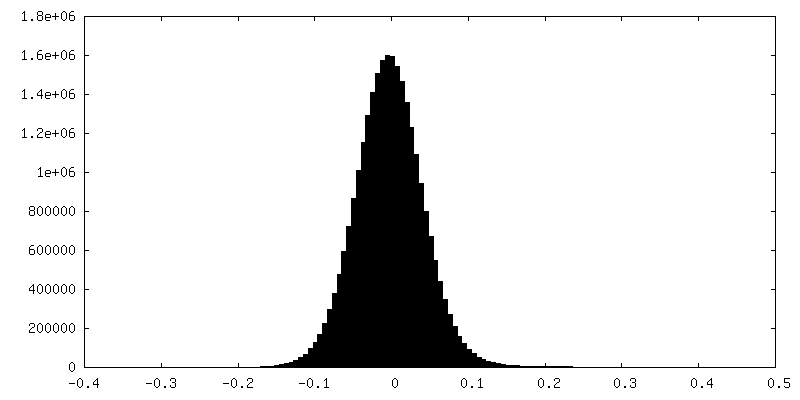
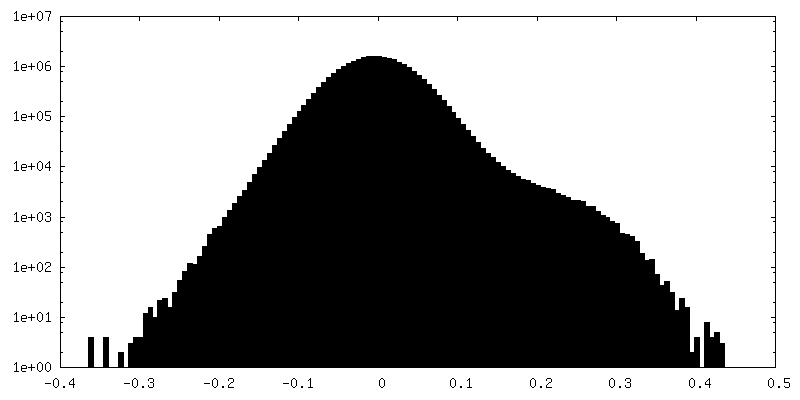

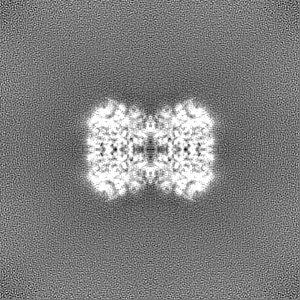




 Sample components
Sample components

 Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN