 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-18311: Compact state - Native eisosome lattice bound to plasma membrane ... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Compact state - Native eisosome lattice bound to plasma membrane microdomain | |||||||||||||||
 Map data Map data | Native eisosome lattice (compact, frame 0) - sharpened map | |||||||||||||||
 Sample Sample |
| |||||||||||||||
 Keywords Keywords | BAR domain / lipid reconstitution / membrane microdomain / LIPID BINDING PROTEIN | |||||||||||||||
| Function / homology |  Function and homology information Function and homology informationprotein localization to eisosome filament / eisosome filament / eisosome assembly / eisosome / lipid droplet / cell periphery / endocytosis / intracellular protein localization / mitochondrial outer membrane / lipid binding ...protein localization to eisosome filament / eisosome filament / eisosome assembly / eisosome / lipid droplet / cell periphery / endocytosis / intracellular protein localization / mitochondrial outer membrane / lipid binding / mitochondrion / plasma membrane / cytoplasm Similarity search - Function | |||||||||||||||
| Biological species |  | |||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.67 Å | |||||||||||||||
 Authors Authors | Kefauver JM / Zou L / Desfosses A / Loewith RJ | |||||||||||||||
| Funding support | European Union,  Switzerland, 4 items Switzerland, 4 items
| |||||||||||||||
 Citation Citation | Journal: Acta Crystallogr D Struct Biol / Year: 2018 Title: Real-space refinement in PHENIX for cryo-EM and crystallography. Authors: Pavel V Afonine / Billy K Poon / Randy J Read / Oleg V Sobolev / Thomas C Terwilliger / Alexandre Urzhumtsev / Paul D Adams /    Abstract: This article describes the implementation of real-space refinement in the phenix.real_space_refine program from the PHENIX suite. The use of a simplified refinement target function enables very fast ...This article describes the implementation of real-space refinement in the phenix.real_space_refine program from the PHENIX suite. The use of a simplified refinement target function enables very fast calculation, which in turn makes it possible to identify optimal data-restraint weights as part of routine refinements with little runtime cost. Refinement of atomic models against low-resolution data benefits from the inclusion of as much additional information as is available. In addition to standard restraints on covalent geometry, phenix.real_space_refine makes use of extra information such as secondary-structure and rotamer-specific restraints, as well as restraints or constraints on internal molecular symmetry. The re-refinement of 385 cryo-EM-derived models available in the Protein Data Bank at resolutions of 6 Å or better shows significant improvement of the models and of the fit of these models to the target maps. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_18311.map.gz | 59.4 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-18311-v30.xmlemd-18311.xml | 22 KB 22 KB | Display Display | EMDB header |
| Images |  emd_18311.png emd_18311.png | 127.3 KB | ||
| Masks | emd_18311_msk_1.map | 64 MB | Mask map | |
| Filedesc metadata | emd-18311.cif.gz | 6.3 KB | ||
| Others | emd_18311_additional_1.map.gzemd_18311_additional_2.map.gzemd_18311_half_map_1.map.gzemd_18311_half_map_2.map.gz | 57.7 MB 32.2 MB 59.4 MB 59.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-18311ftp://ftp.pdbj.org/pub/emdb/structures/EMD-18311 http://ftp.pdbj.org/pub/emdb/structures/EMD-18311ftp://ftp.pdbj.org/pub/emdb/structures/EMD-18311 | HTTPS FTP |
-Validation report
| Summary document | emd_18311_validation.pdf.gz | 1 MB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_18311_full_validation.pdf.gz | 1 MB | Display | |
| Data in XML | emd_18311_validation.xml.gz | 12.1 KB | Display | |
| Data in CIF | emd_18311_validation.cif.gz | 14.2 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-18311ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-18311 | HTTPS FTP |
-Related structure data
| Related structure data |  8qbeMC  8qbfMC  8qb7C  8qb8C  8qb9C  8qbbC  8qbdC  8qbgC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_18311.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Native eisosome lattice (compact, frame 0) - sharpened map | ||||||||||||||||||||||||||||||||||||
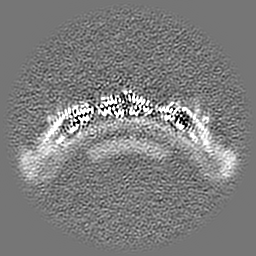
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.327 Å | ||||||||||||||||||||||||||||||||||||
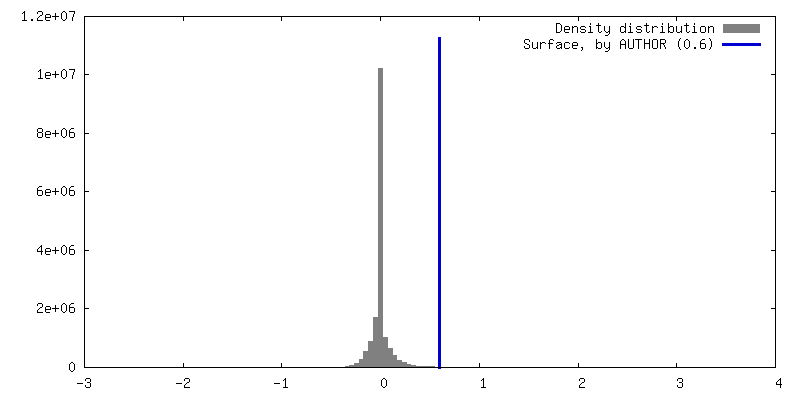
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-Supplemental data
-Mask #1
| File | emd_18311_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
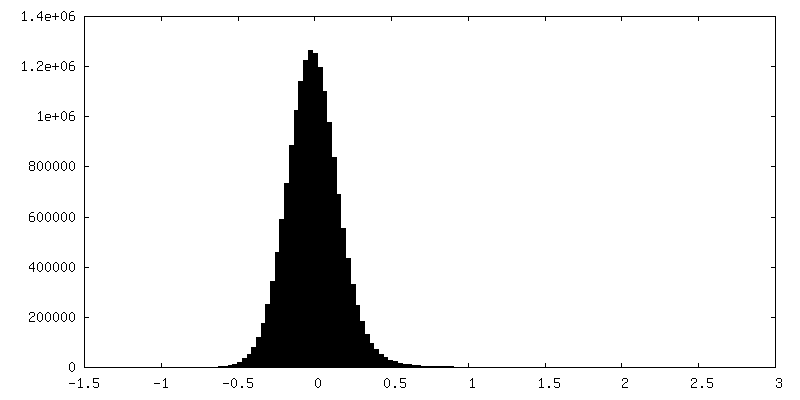
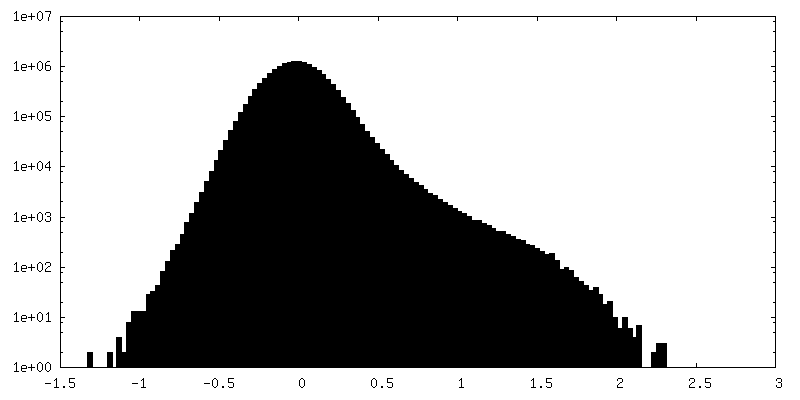
| Density Histograms |






-Additional map: Native eisosome lattice (compact, frame 0) - deepEMhancer...
| File | emd_18311_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Native eisosome lattice (compact, frame 0) - deepEMhancer sharpened map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







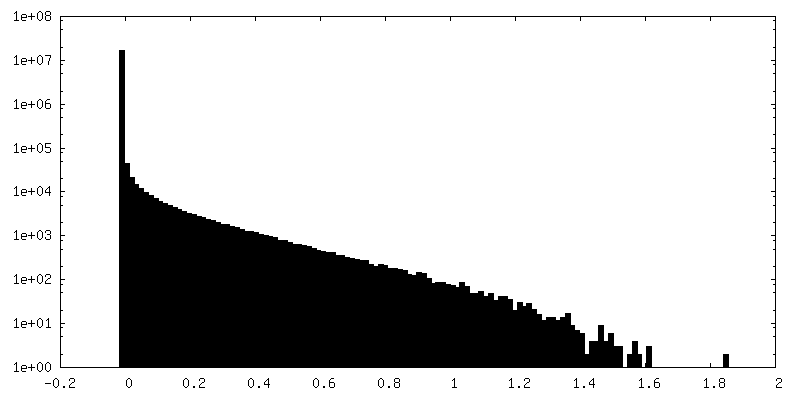
-Additional map: Native eisosome lattice (compact, frame 0) - unsharpened map
| File | emd_18311_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Native eisosome lattice (compact, frame 0) - unsharpened map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |


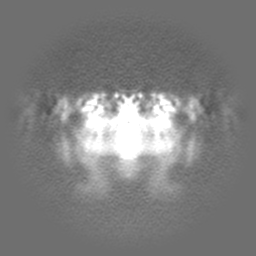
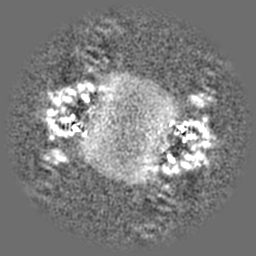
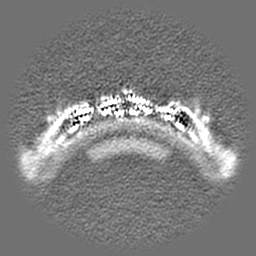
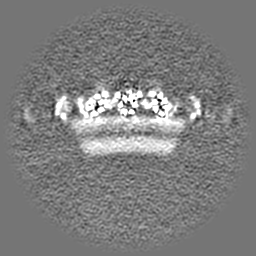
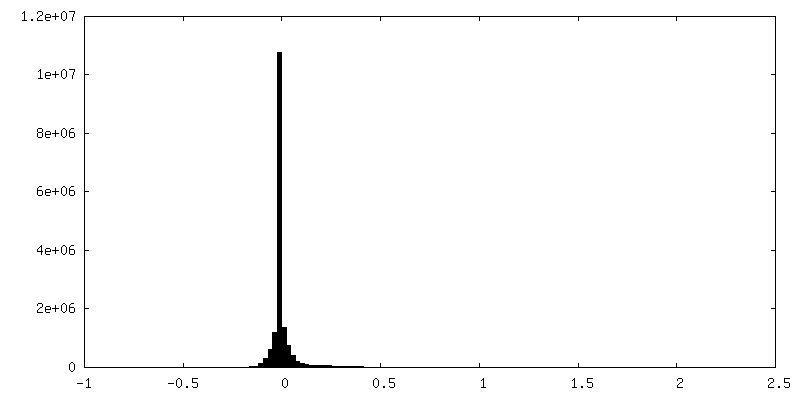
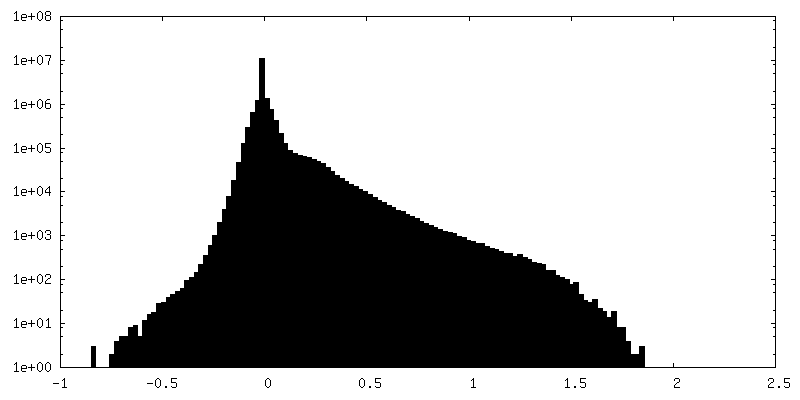
-Half map: Native eisosome lattice (compact, frame 0) - half map A
| File | emd_18311_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Native eisosome lattice (compact, frame 0) - half map A | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |
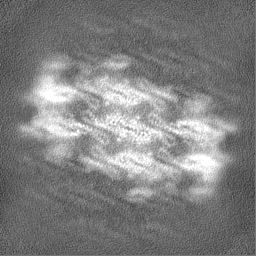

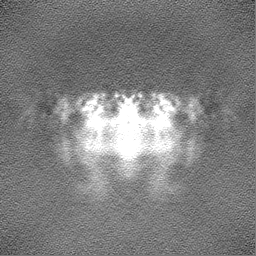

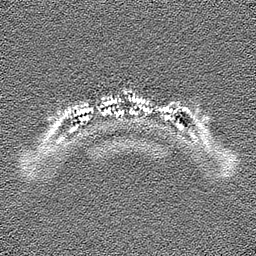
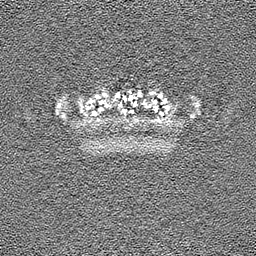

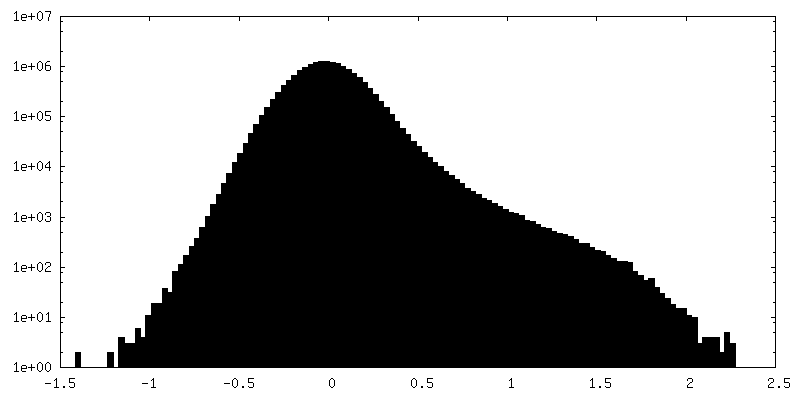
-Half map: Native eisosome lattice (compact, frame 0) - half map B
| File | emd_18311_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Native eisosome lattice (compact, frame 0) - half map B | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |


- Sample components
Sample components
-Entire : Compact state - Helical lattice of native Pil1/Lsp1 protein bound...
| Entire | Name: Compact state - Helical lattice of native Pil1/Lsp1 protein bound to plasma membrane microdomain |
|---|---|
| Components |
|
-Supramolecule #1: Compact state - Helical lattice of native Pil1/Lsp1 protein bound...
| Supramolecule | Name: Compact state - Helical lattice of native Pil1/Lsp1 protein bound to plasma membrane microdomain type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: |
-Macromolecule #1: Sphingolipid long chain base-responsive protein PIL1
| Macromolecule | Name: Sphingolipid long chain base-responsive protein PIL1 / type: protein_or_peptide / ID: 1 / Number of copies: 14 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 38.393043 KDa |
| Sequence | String: MHRTYSLRNS RAPTASQLQN PPPPPSTTKG RFFGKGGLAY SFRRSAAGAF GPELSRKLSQ LVKIEKNVLR SMELTANERR DAAKQLSIW GLENDDDVSD ITDKLGVLIY EVSELDDQFI DRYDQYRLTL KSIRDIEGSV QPSRDRKDKI TDKIAYLKYK D PQSPKIEV ...String: MHRTYSLRNS RAPTASQLQN PPPPPSTTKG RFFGKGGLAY SFRRSAAGAF GPELSRKLSQ LVKIEKNVLR SMELTANERR DAAKQLSIW GLENDDDVSD ITDKLGVLIY EVSELDDQFI DRYDQYRLTL KSIRDIEGSV QPSRDRKDKI TDKIAYLKYK D PQSPKIEV LEQELVRAEA ESLVAEAQLS NITRSKLRAA FNYQFDSIIE HSEKIALIAG YGKALLELLD DSPVTPGETR PA YDGYEAS KQIIIDAESA LNEWTLDSAQ VKPTLSFKQD YEDFEPEEGE EEEEEDGQGR WSEDEQEDGQ IEEPEQEEEG AVE EHEQVG HQQSESLPQQ TTA UniProtKB: Sphingolipid long chain base-responsive protein PIL1 |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | helical array |
-Sample preparation
| Buffer | pH: 7 / Details: 50mM PIPES pH 7, 300mM NaCl, 1mM CHAPS, 0.5mM DTT |
|---|---|
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 90 % / Chamber temperature: 291 K / Instrument: LEICA EM GP |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 QUANTUM (4k x 4k) / Average electron dose: 40.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 1.8 µm / Nominal defocus min: 0.8 µm |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Startup model | Type of model: NONE |
|---|---|
| Final reconstruction | Resolution.type: BY AUTHOR / Resolution: 3.67 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: cryoSPARC (ver. 4.1.2) / Number images used: 63118 |
| Initial angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.0.8) |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD / Software - Name: cryoSPARC (ver. 4.1.2) |
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Source name: AlphaFold / Chain - Initial model type: in silico model |
|---|---|
| Refinement | Space: REAL |
| Output model | PDB-8qbe: PDB-8qbf: |

