Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-17944: Cryo-EM structure of an asymmetric nucleosome (10-N-0) from a Gla... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of an asymmetric nucleosome (10-N-0) from a Glacios/Falcon3EC | |||||||||
 Map data Map data | Map from homogeneous refinement in cryosparc. | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Nucleosome / NCP / DNA / Histones / DNA BINDING PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationstructural constituent of chromatin / nucleosome / nucleosome assembly / heterochromatin formation / protein heterodimerization activity / DNA binding / nucleoplasm / nucleus Similarity search - Function | |||||||||
| Biological species | ||||||||||
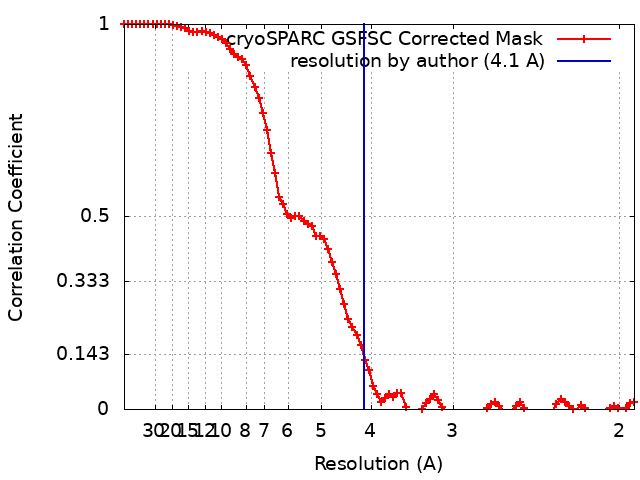
| Method | single particle reconstruction / cryo EM / Resolution: 4.1 Å | |||||||||
 Authors Authors | Gaullier G | |||||||||
| Funding support | 1 items
| |||||||||
 Citation Citation | Journal: Elife / Year: 2021 Title: Structure and dynamics of the chromatin remodeler ALC1 bound to a PARylated nucleosome. Authors: Luka Bacic / Guillaume Gaullier / Anton Sabantsev / Laura C Lehmann / Klaus Brackmann / Despoina Dimakou / Mario Halic / Graeme Hewitt / Simon J Boulton / Sebastian Deindl /    Abstract: The chromatin remodeler ALC1 is recruited to and activated by DNA damage-induced poly(ADP-ribose) (PAR) chains deposited by PARP1/PARP2/HPF1 upon detection of DNA lesions. ALC1 has emerged as a ...The chromatin remodeler ALC1 is recruited to and activated by DNA damage-induced poly(ADP-ribose) (PAR) chains deposited by PARP1/PARP2/HPF1 upon detection of DNA lesions. ALC1 has emerged as a candidate drug target for cancer therapy as its loss confers synthetic lethality in homologous recombination-deficient cells. However, structure-based drug design and molecular analysis of ALC1 have been hindered by the requirement for PARylation and the highly heterogeneous nature of this post-translational modification. Here, we reconstituted an ALC1 and PARylated nucleosome complex modified in vitro using PARP2 and HPF1. This complex was amenable to cryo-EM structure determination without cross-linking, which enabled visualization of several intermediate states of ALC1 from the recognition of the PARylated nucleosome to the tight binding and activation of the remodeler. Functional biochemical assays with PARylated nucleosomes highlight the importance of nucleosomal epitopes for productive remodeling and suggest that ALC1 preferentially slides nucleosomes away from DNA breaks. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_17944.map.gz | 31.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-17944-v30.xmlemd-17944.xml | 24.6 KB 24.6 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_17944_fsc.xml | 8.5 KB | Display | FSC data file |
| Images | 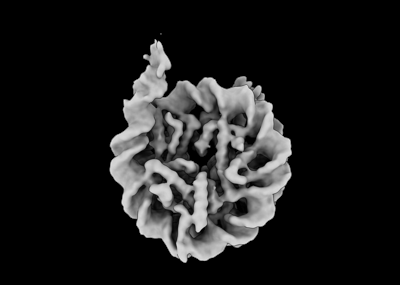 emd_17944.png emd_17944.png | 36.3 KB | ||
| Masks | emd_17944_msk_1.mapemd_17944_msk_2.map | 64 MB 64 MB | Mask map | |
| Filedesc metadata | emd-17944.cif.gz | 6.3 KB | ||
| Others | emd_17944_additional_1.map.gzemd_17944_half_map_1.map.gzemd_17944_half_map_2.map.gz | 57.5 MB 59.5 MB 59.5 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-17944ftp://ftp.pdbj.org/pub/emdb/structures/EMD-17944 http://ftp.pdbj.org/pub/emdb/structures/EMD-17944ftp://ftp.pdbj.org/pub/emdb/structures/EMD-17944 | HTTPS FTP |
-Related structure data



-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_17944.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Map from homogeneous refinement in cryosparc. | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.952 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_17944_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Mask #2
| File | emd_17944_msk_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Additional map: Map after post-processing with deepEMhancer, using the "tighTarget"...
| File | emd_17944_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Map after post-processing with deepEMhancer, using the "tighTarget" weights. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Half map: Half-map A from homogeneous refinement in cryosparc.
| File | emd_17944_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half-map A from homogeneous refinement in cryosparc. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: Half-map B from homogeneous refinement in cryosparc.
| File | emd_17944_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half-map B from homogeneous refinement in cryosparc. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






- Sample components
Sample components
-Entire : 601 nucleosome with 10 additional bp
| Entire | Name: 601 nucleosome with 10 additional bp |
|---|---|
| Components |
|
-Supramolecule #1: 601 nucleosome with 10 additional bp
| Supramolecule | Name: 601 nucleosome with 10 additional bp / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all Details: The histones were recombinantly expressed and purified. The DNA was prepared by PCR. The nucleosomes were assembled by salt gradient dialysis. |
|---|---|
| Molecular weight | Theoretical: 205.8 KDa |
-Macromolecule #1: Histone H3.2
| Macromolecule | Name: Histone H3.2 / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism:  |
| Sequence | String: MARTKQTARK STGGKAPRKQ LATKAARKSA PATGGVKKPH RYRPGTVALR EIRRYQKSTE LLIRKLPFQR LVREIAQDFK TDLRFQSSAV MALQEASEAY LVALFEDTNL AAIHAKRVTI MPKDIQLARR IRGERA UniProtKB: Histone H3.2 |
-Macromolecule #2: Histone H4
| Macromolecule | Name: Histone H4 / type: protein_or_peptide / ID: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism: |
| Sequence | String: MSGRGKGGKG LGKGGAKRHR KVLRDNIQGI TKPAIRRLAR RGGVKRISGL IYEETRGVLK VFLENVIRDA VTYTEHAKRK TVTAMDVVYA LKRQGRTLYG FGG UniProtKB: Histone H4 |
-Macromolecule #3: Histone H2A type 1
| Macromolecule | Name: Histone H2A type 1 / type: protein_or_peptide / ID: 3 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism: |
| Sequence | String: MSGRGKQGGK TRAKAKTRSS RAGLQFPVGR VHRLLRKGNY AERVGAGAPV YLAAVLEYLT AEILELAGNA ARDNKKTRII PRHLQLAVRN DEELNKLLGR VTIAQGGVLP NIQSVLLPKK TESSKSAKSK UniProtKB: Histone H2A type 1 |
-Macromolecule #4: Histone H2B 1.1
| Macromolecule | Name: Histone H2B 1.1 / type: protein_or_peptide / ID: 4 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism: |
| Sequence | String: MAKSAPAPKK GSKKAVTKTQ KKDGKKRRKT RKESYAIYVY KVLKQVHPDT GISSKAMSIM NSFVNDVFER IAGEASRLAH YNKRSTITSR EIQTAVRLLL PGELAKHAVS EGTKAVTKYT SAK UniProtKB: Histone H2B 1.1 |
-Macromolecule #5: Widom 601 sequence with 10 additional bp on the strong side
| Macromolecule | Name: Widom 601 sequence with 10 additional bp on the strong side type: dna / ID: 5 / Classification: DNA |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Sequence | String: AATCGATGTA TATATCTGAC ACGTGCCTGG AGACTAGGGA GTAATCCCCT TGGCGGTTAA AACGCGGGGG ACAGCGCGTA CGTGCGTTTA AGCGGTGCTA GAGCTGTCTA CGACCAATTG AGCGGCCTCG GCACCGGGAT TCTGATGGTC ACCTAGA |
-Macromolecule #6: Widom 601 sequence with 10 additional bp on the strong side
| Macromolecule | Name: Widom 601 sequence with 10 additional bp on the strong side type: dna / ID: 6 / Classification: DNA |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Sequence | String: TCTAGGTGAC CATCAGAATC CCGGTGCCGA GGCCGCTCAA TTGGTCGTAG ACAGCTCTAG CACCGCTTAA ACGCACGTAC GCGCTGTCCC CCGCGTTTTA ACCGCCAAGG GGATTACTCC CTAGTCTCCA GGCACGTGTC AGATATATAC ATCGATT |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.17 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 8 Component:
| |||||||||
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 120 sec. / Pretreatment - Atmosphere: AIR / Pretreatment - Pressure: 0.04 kPa / Details: Pelco easiGlow operated with a 30 mA current. | |||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV Details: Blot time 4s Blot force 0 4 uL of sample were applied on the grid (2 uL on each side of the grid). | |||||||||
| Details | At the time of vitrification, the nucleosome sample used had spent about one year stored at 4C (this increases the propensity of nucleosomes to unwrap upon vitrification). |
- Electron microscopy
Electron microscopy
| Microscope | TFS GLACIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Detector mode: COUNTING / Number grids imaged: 1 / Number real images: 1537 / Average electron dose: 40.01 e/Å2 / Details: 40 frames per movie |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 0.5 µm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |