 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-17360: Cryo-EM structure of the anaerobic ribonucleotide reductase from ... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of the anaerobic ribonucleotide reductase from Prevotella copri in its tetrameric state produced in the presence of dATP and CTP | |||||||||||||||
 Map data Map data | Map post-processed using DeepEMhancer | |||||||||||||||
 Sample Sample |
| |||||||||||||||
 Keywords Keywords | ribonucleotide reductase glycyl radical enzyme allosteric regulation nucleotide biosynthesis / OXIDOREDUCTASE / BIOSYNTHETIC PROTEIN | |||||||||||||||
| Function / homology | Ribonucleoside-triphosphate reductase, anaerobic / Anaerobic ribonucleoside-triphosphate reductase / ribonucleoside-triphosphate reductase (thioredoxin) activity / ATP-cone domain / ATP cone domain / ATP-cone domain profile. / DNA replication / ATP binding / Anaerobic ribonucleoside-triphosphate reductase Function and homology information Function and homology information | |||||||||||||||
| Biological species |  Prevotella copri (bacteria) / Segatella copri (bacteria) Prevotella copri (bacteria) / Segatella copri (bacteria) | |||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.59 Å | |||||||||||||||
 Authors Authors | Banerjee I / Bimai O / Sjoberg BM / Logan DT | |||||||||||||||
| Funding support |  Sweden, Sweden,  United States, 4 items United States, 4 items
| |||||||||||||||
 Citation Citation | Journal: Elife / Year: 2024 Title: Nucleotide binding to the ATP-cone in anaerobic ribonucleotide reductases allosterically regulates activity by modulating substrate binding. Authors: Ornella Bimai / Ipsita Banerjee / Inna Rozman Grinberg / Ping Huang / Lucas Hultgren / Simon Ekström / Daniel Lundin / Britt-Marie Sjöberg / Derek T Logan / Abstract: A small, nucleotide-binding domain, the ATP-cone, is found at the N-terminus of most ribonucleotide reductase (RNR) catalytic subunits. By binding adenosine triphosphate (ATP) or deoxyadenosine ...A small, nucleotide-binding domain, the ATP-cone, is found at the N-terminus of most ribonucleotide reductase (RNR) catalytic subunits. By binding adenosine triphosphate (ATP) or deoxyadenosine triphosphate (dATP) it regulates the enzyme activity of all classes of RNR. Functional and structural work on aerobic RNRs has revealed a plethora of ways in which dATP inhibits activity by inducing oligomerisation and preventing a productive radical transfer from one subunit to the active site in the other. Anaerobic RNRs, on the other hand, store a stable glycyl radical next to the active site and the basis for their dATP-dependent inhibition is completely unknown. We present biochemical, biophysical, and structural information on the effects of ATP and dATP binding to the anaerobic RNR from . The enzyme exists in a dimer-tetramer equilibrium biased towards dimers when two ATP molecules are bound to the ATP-cone and tetramers when two dATP molecules are bound. In the presence of ATP, NrdD is active and has a fully ordered glycyl radical domain (GRD) in one monomer of the dimer. Binding of dATP to the ATP-cone results in loss of activity and increased dynamics of the GRD, such that it cannot be detected in the cryo-EM structures. The glycyl radical is formed even in the dATP-bound form, but the substrate does not bind. The structures implicate a complex network of interactions in activity regulation that involve the GRD more than 30 Å away from the dATP molecules, the allosteric substrate specificity site and a conserved but previously unseen flap over the active site. Taken together, the results suggest that dATP inhibition in anaerobic RNRs acts by increasing the flexibility of the flap and GRD, thereby preventing both substrate binding and radical mobilisation. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_17360.map.gz | 318 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-17360-v30.xmlemd-17360.xml | 25.1 KB 25.1 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_17360_fsc.xml | 14.7 KB | Display | FSC data file |
| Images |  emd_17360.png emd_17360.png | 66.5 KB | ||
| Masks | emd_17360_msk_1.map | 343 MB | Mask map | |
| Filedesc metadata | emd-17360.cif.gz | 7.3 KB | ||
| Others | emd_17360_additional_1.map.gzemd_17360_half_map_1.map.gzemd_17360_half_map_2.map.gz | 317.5 MB 318.1 MB 318.1 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-17360ftp://ftp.pdbj.org/pub/emdb/structures/EMD-17360 http://ftp.pdbj.org/pub/emdb/structures/EMD-17360ftp://ftp.pdbj.org/pub/emdb/structures/EMD-17360 | HTTPS FTP |
-Validation report
| Summary document | emd_17360_validation.pdf.gz | 717.9 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_17360_full_validation.pdf.gz | 717.5 KB | Display | |
| Data in XML | emd_17360_validation.xml.gz | 23.8 KB | Display | |
| Data in CIF | emd_17360_validation.cif.gz | 31.4 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-17360ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-17360 | HTTPS FTP |
-Related structure data
| Related structure data |  8p2cMC  8p23C  8p27C  8p28C  8p2dC  8p2sC  8p39C C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_17360.map.gz / Format: CCP4 / Size: 343 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Map post-processed using DeepEMhancer | ||||||||||||||||||||||||||||||||||||
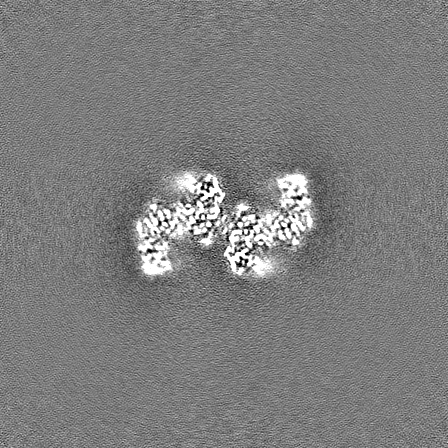
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.8464 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) X (Row.)
X (Row.) Y (Col.)
Y (Col.)




















-Supplemental data
-Mask #1
| File | emd_17360_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
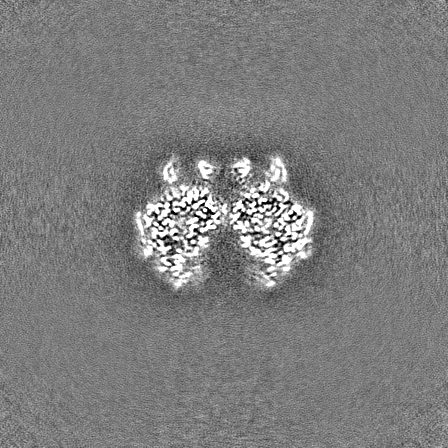
| Projections & Slices |
| ||||||||||||
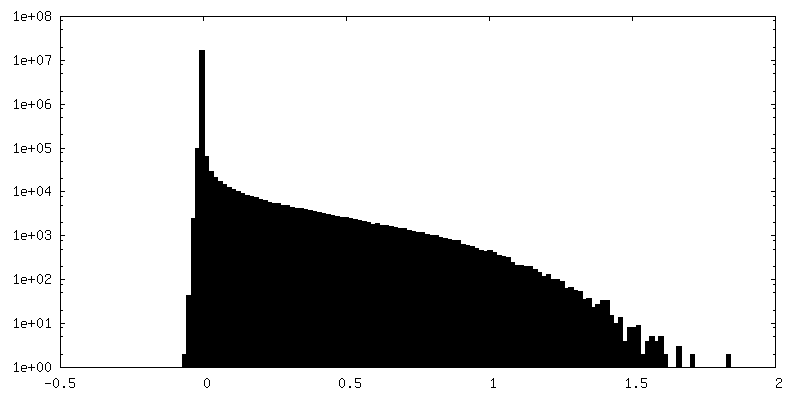
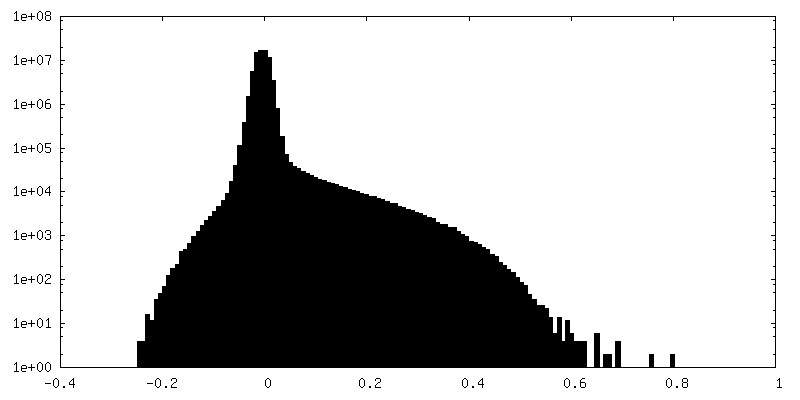
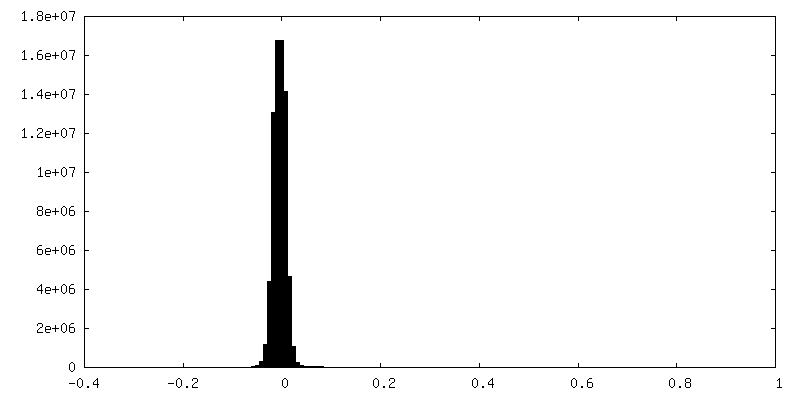
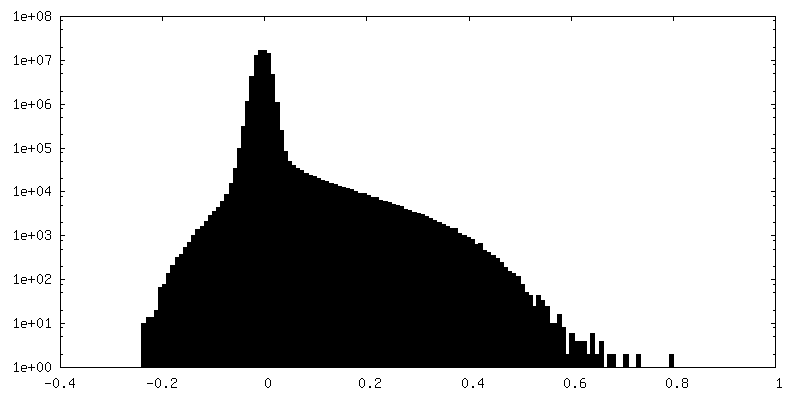
| Density Histograms |






-Additional map: Unsharpened map from non-uniform refinement in cryoSPARC
| File | emd_17360_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unsharpened map from non-uniform refinement in cryoSPARC | ||||||||||||
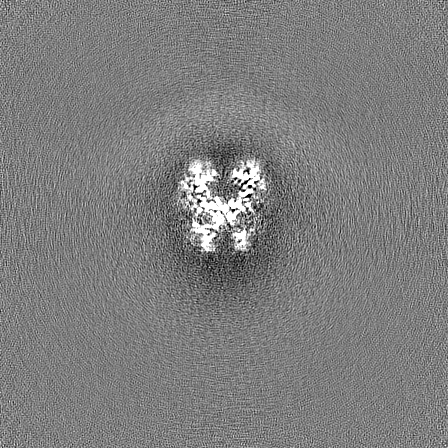
| Projections & Slices |
| ||||||||||||
| Density Histograms |






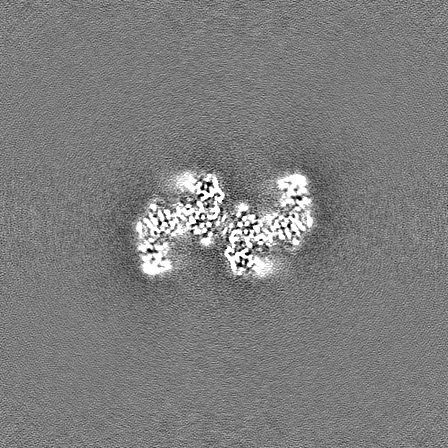
-Half map: Half map A from non-uniform refinement in cryoSPARC
| File | emd_17360_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map A from non-uniform refinement in cryoSPARC | ||||||||||||
| Projections & Slices |
| ||||||||||||
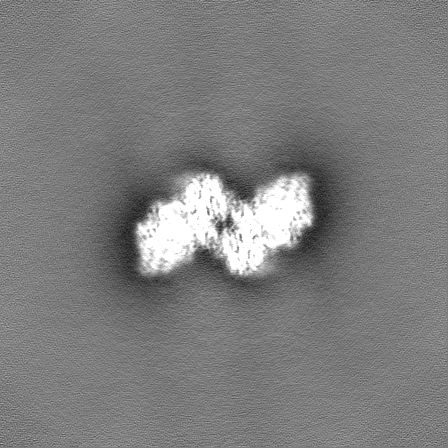
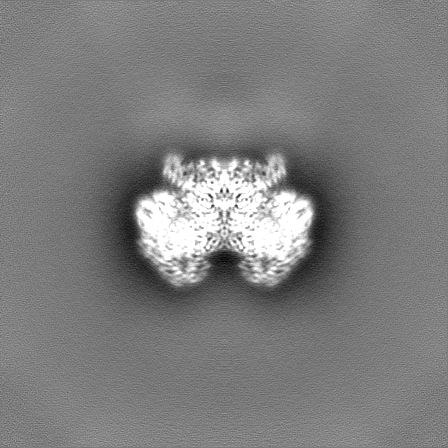
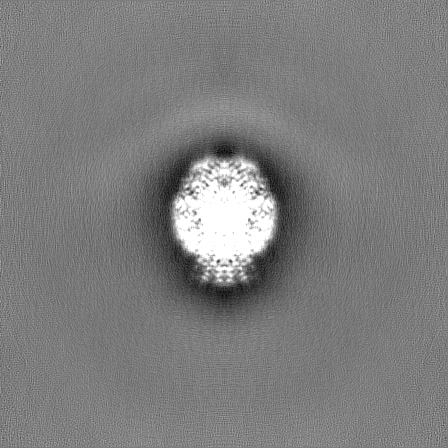
| Density Histograms |
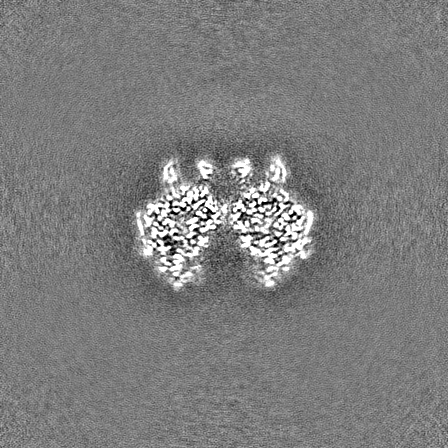
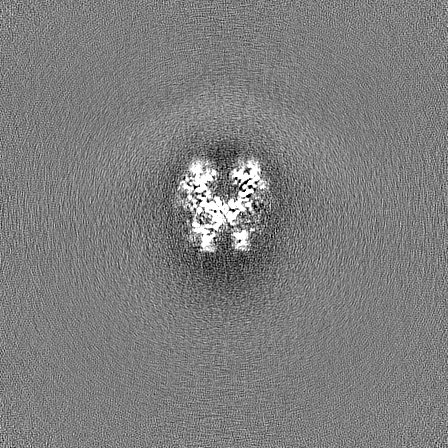
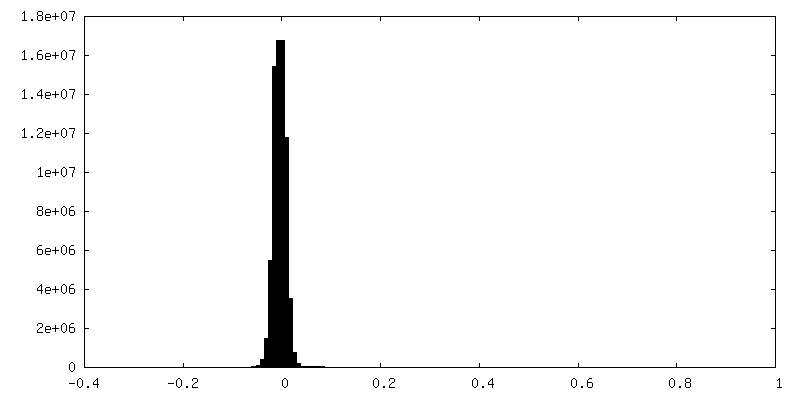
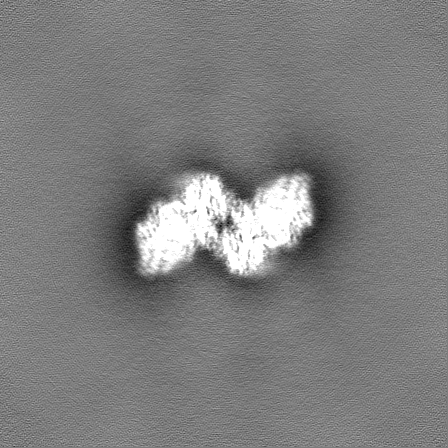
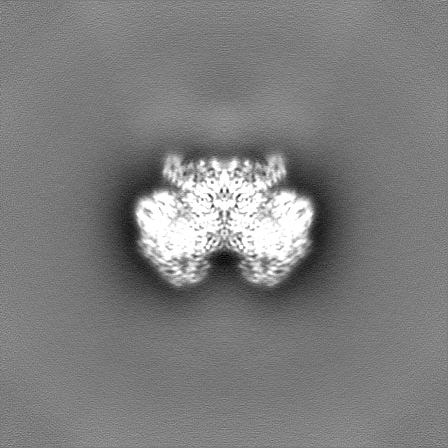
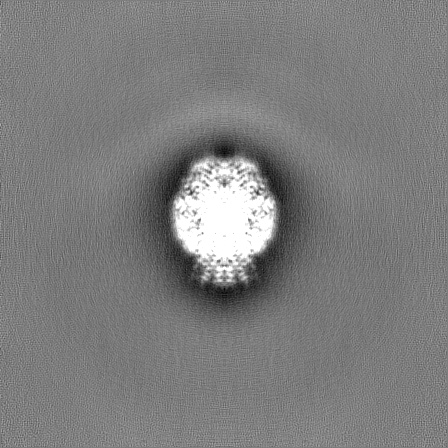
-Half map: Half map B from non-uniform refinement in cryoSPARC
| File | emd_17360_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map B from non-uniform refinement in cryoSPARC | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |
- Sample components
Sample components
-Entire : Anaerobic ribonucleotide reductase from Prevotella copri in its t...
| Entire | Name: Anaerobic ribonucleotide reductase from Prevotella copri in its tetrameric, dATP/CTP-bound state |
|---|---|
| Components |
|
-Supramolecule #1: Anaerobic ribonucleotide reductase from Prevotella copri in its t...
| Supramolecule | Name: Anaerobic ribonucleotide reductase from Prevotella copri in its tetrameric, dATP/CTP-bound state type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: Prevotella copri (bacteria) |
| Molecular weight | Theoretical: 336.8 KDa |
-Macromolecule #1: Anaerobic ribonucleoside-triphosphate reductase
| Macromolecule | Name: Anaerobic ribonucleoside-triphosphate reductase / type: protein_or_peptide / ID: 1 / Number of copies: 4 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Segatella copri (bacteria) |
| Molecular weight | Theoretical: 84.636055 KDa |
| Recombinant expression | Organism: |
| Sequence | String: GPGSMIQTVV KRDGRIVGFN EQKIMAAIRK AMLHTDKGED TTLIEQITDH ISYRGKSQMS VEAIQDAIEM ELMKSARKDV AQKYIAYRN QRNIARKAKT RDVFMSIVNA KNNDITRENA NMNADTPAGM MMKFASETTK PFVDDYLLSE DVRDAVMHNY I HIHDKDYY ...String: GPGSMIQTVV KRDGRIVGFN EQKIMAAIRK AMLHTDKGED TTLIEQITDH ISYRGKSQMS VEAIQDAIEM ELMKSARKDV AQKYIAYRN QRNIARKAKT RDVFMSIVNA KNNDITRENA NMNADTPAGM MMKFASETTK PFVDDYLLSE DVRDAVMHNY I HIHDKDYY PTKSLTCVQH PLDVILNHGF TAGHGSSRPA KRIETAAVLA CISLETCQNE MHGGQAIPAF DFYLAPYVRM SY QEEVKNL EKLTGEDLSN LYDAPIDDYI EKPLDGLQGR ERLEQHAINK TVNRVHQAME AFIHNMNTIH SRGGNQVVFS SIN YGTDTS AEGRCIMREI LQSTYQGVGN GETAIFPIQI WKKKRGVNYL PEDRNYDLYK LACKVTARRF FPNFLNLDAT FNQN EKWRA DDPERYKWEI ATMGCRTRVF EDRWGEKTSI ARGNLSFSTI NIVKLAIECM GIENEKQRID MFFAKLDNIL DITAK QLDE RFQFQKTAMA KQFPLLMKYL WVGAENLKPE ETIESVINHG TLGIGFIGLA ECLVALIGKH HGESEKAQEL GLKIIT YMR DRANEFSEQY HHNYSILATP AEGLSGKFTK KDRKQFGVIP GVTDRDYYTN SNHVPVYYKC TALKKAQIEA PYHDLTR GG HIFYVEIDGD ATHNPSVIES VVDMMDKYNM GYGSVNHNRN RCLDCGYENA DAHLEVCPKC GSHHIDKLQR ITGYLVGT T DRWNSGKLAE LHDRVTHIGG EK UniProtKB: Anaerobic ribonucleoside-triphosphate reductase |
-Macromolecule #2: 2'-DEOXYADENOSINE 5'-TRIPHOSPHATE
| Macromolecule | Name: 2'-DEOXYADENOSINE 5'-TRIPHOSPHATE / type: ligand / ID: 2 / Number of copies: 12 / Formula: DTP |
|---|---|
| Molecular weight | Theoretical: 491.182 Da |
| Chemical component information |  ChemComp-DTP: |
-Macromolecule #3: MAGNESIUM ION
| Macromolecule | Name: MAGNESIUM ION / type: ligand / ID: 3 / Number of copies: 4 / Formula: MG |
|---|---|
| Molecular weight | Theoretical: 24.305 Da |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.5 mg/mL | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| |||||||||||||||||||||
| Grid | Model: Quantifoil R2/1 / Material: COPPER / Mesh: 300 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 60 sec. / Pretreatment - Atmosphere: OTHER | |||||||||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV / Details: blot force 1, 5s blot time. |
- Electron microscopy
Electron microscopy
| Microscope | TFS KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Bioquantum |
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Digitization - Dimensions - Width: 5760 pixel / Digitization - Dimensions - Height: 4092 pixel / Number grids imaged: 1 / Number real images: 11780 / Average exposure time: 2.0 sec. / Average electron dose: 38.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: SPOT SCAN / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.0 µm / Nominal defocus min: 0.6 µm / Nominal magnification: 105000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
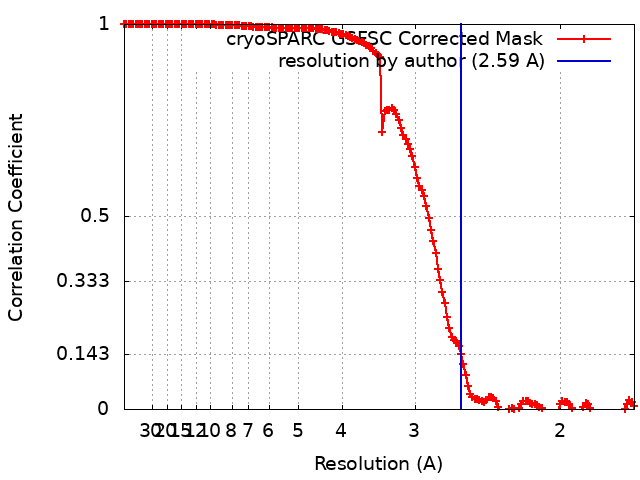
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Details | Manual fitting was done using Coot and automatic real space refinement used phenix.refine |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Target criteria: correlation coefficient |
| Output model | PDB-8p2c: |
