Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-17195: CryoEM reconstruction of a TRAP protein cage made out of 12 11-me... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CryoEM reconstruction of a TRAP protein cage made out of 12 11-membered rings | |||||||||
 Map data Map data | main map | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | protein cage / TRAP / VIRUS LIKE PARTICLE | |||||||||
| Function / homology | Transcription attenuation protein MtrB / Tryptophan RNA-binding attenuator protein domain / Tryptophan RNA-binding attenuator protein / Tryptophan RNA-binding attenuator protein-like domain superfamily / DNA-templated transcription termination / regulation of DNA-templated transcription / RNA binding / Transcription attenuation protein MtrB Function and homology information Function and homology information | |||||||||
| Biological species |   Geobacillus stearothermophilus (bacteria) Geobacillus stearothermophilus (bacteria) | |||||||||
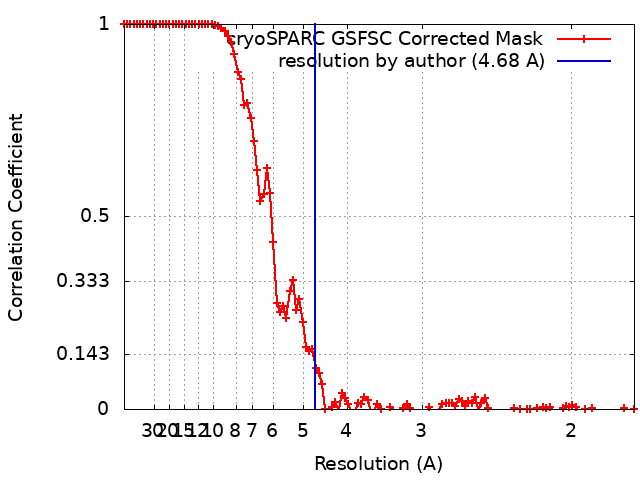
| Method | single particle reconstruction / cryo EM / Resolution: 4.68 Å | |||||||||
 Authors Authors | Biela AP | |||||||||
| Funding support |  Poland, 1 items Poland, 1 items
| |||||||||
 Citation Citation | Journal: J Mater Chem B / Year: 2024 Title: An artificial protein cage made from a 12-membered ring. Authors: Izabela Stupka / Artur P Biela / Bernard Piette / Agnieszka Kowalczyk / Karolina Majsterkiewicz / Kinga Borzęcka-Solarz / Antonina Naskalska / Jonathan G Heddle /  Abstract: Artificial protein cages have great potential in diverse fields including as vaccines and drug delivery vehicles. TRAP-cage is an artificial protein cage notable for the way in which the interface ...Artificial protein cages have great potential in diverse fields including as vaccines and drug delivery vehicles. TRAP-cage is an artificial protein cage notable for the way in which the interface between its ring-shaped building blocks can be modified such that the conditions under which cages disassemble can be controlled. To date, TRAP-cages have been constructed from homo-11mer rings, , hendecamers. This is interesting as convex polyhedra with identical regular faces cannot be formed from hendecamers. TRAP-cage overcomes this limitation due to intrinsic flexibility, allowing slight deformation to absorb any error. The resulting TRAP-cage made from 24 TRAP 11mer rings is very close to regular with only very small errors necessary to allow the cage to form. The question arises as to the limits of the error that can be absorbed by a protein structure in this way before the formation of an apparently regular convex polyhedral becomes impossible. Here we use a naturally occurring TRAP variant consisting of twelve identical monomers (, a dodecamer) to probe these limits. We show that it is able to form an apparently regular protein cage consisting of twelve TRAP rings. Comparison of the cryo-EM structure of the new cage with theoretical models and related cages gives insight into the rules of cage formation and allows us to predict other cages that may be formed given TRAP-rings consisting of different numbers of monomers. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_17195.map.gz | 117.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-17195-v30.xmlemd-17195.xml | 15.2 KB 15.2 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_17195_fsc.xml | 13.8 KB | Display | FSC data file |
| Images |  emd_17195.png emd_17195.png | 66.1 KB | ||
| Filedesc metadata | emd-17195.cif.gz | 4.8 KB | ||
| Others | emd_17195_half_map_1.map.gzemd_17195_half_map_2.map.gz | 114.2 MB 114.2 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-17195ftp://ftp.pdbj.org/pub/emdb/structures/EMD-17195 http://ftp.pdbj.org/pub/emdb/structures/EMD-17195ftp://ftp.pdbj.org/pub/emdb/structures/EMD-17195 | HTTPS FTP |
-Related structure data
| Related structure data | C: citing same article ( |
|---|---|
| Similar structure data |

-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_17195.map.gz / Format: CCP4 / Size: 125 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | main map | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.86 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Half map: half map A
| File | emd_17195_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map A | ||||||||||||
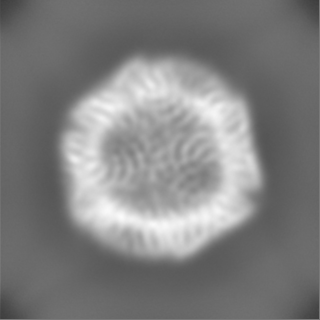
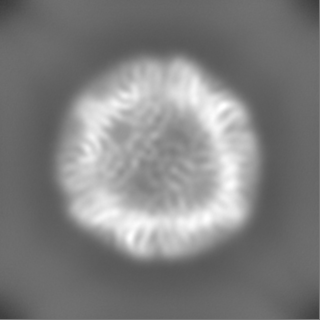
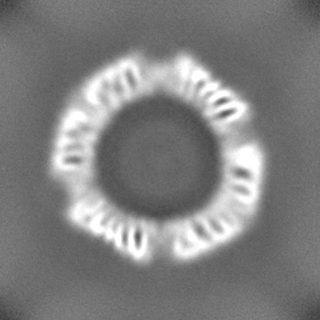
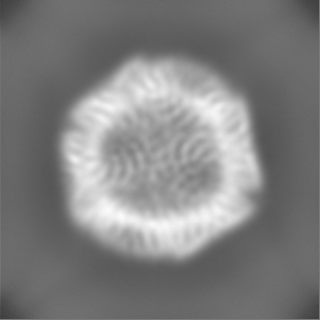
| Projections & Slices |
| ||||||||||||
| Density Histograms |



-Half map: half map B
| File | emd_17195_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map B | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |

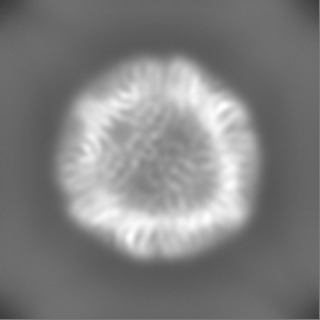



- Sample components
Sample components
-Entire : protein cage composed of 12 identical 11-membered TRAP rings
| Entire | Name: protein cage composed of 12 identical 11-membered TRAP rings |
|---|---|
| Components |
|
-Supramolecule #1: protein cage composed of 12 identical 11-membered TRAP rings
| Supramolecule | Name: protein cage composed of 12 identical 11-membered TRAP rings type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Geobacillus stearothermophilus (bacteria) |
| Molecular weight | Theoretical: 1.2 MDa |
-Macromolecule #1: Transcription attenuation protein MtrB
| Macromolecule | Name: Transcription attenuation protein MtrB / type: protein_or_peptide / ID: 1 / Details: point mutations at position 35 and 64 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Geobacillus stearothermophilus (bacteria) |
| Recombinant expression | Organism: |
| Sequence | String: MYTNSDFVVI KALEDGVNVI GLTRGADTRF HHSECLDKGE VLIAQFTEHT SAIKVRGKAY IQTSHGVIES EGKK UniProtKB: Transcription attenuation protein MtrB |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1 mg/mL |
|---|---|
| Buffer | pH: 7.4 |
| Grid | Model: Quantifoil R2/1 / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 70 sec. / Pretreatment - Atmosphere: AIR |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Average electron dose: 40.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 3.5 µm / Nominal defocus min: 1.2 µm |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |