ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Automated simulation-based refinement of maltoporin into a cryo-EM density | |||||||||
 マップデータ マップデータ | Half map 2, box 128 | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード | maltoporin / density fit / automated refinement / cryo-em / MEMBRANE PROTEIN | |||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報maltodextrin transmembrane transporter activity / maltose transporting porin activity / pore complex / monoatomic ion transport / cell outer membrane 類似検索 - 分子機能 | |||||||||
| 生物種 |  | |||||||||
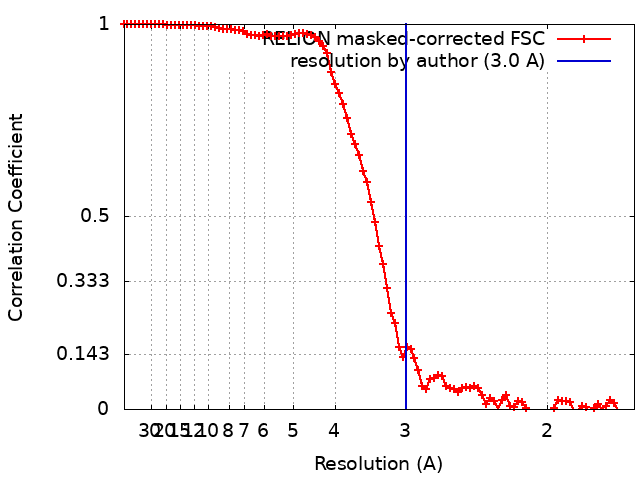
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 3.0 Å | |||||||||
 データ登録者 データ登録者 | Yvonnesdotter L / Rovsnik U / Blau C / Lycksell M / Howard RJ / Lindahl E | |||||||||
| 資金援助 |  スウェーデン, 2件 スウェーデン, 2件
| |||||||||
 引用 引用 | ジャーナル: Biophys J / 年: 2023 タイトル: Automated simulation-based membrane protein refinement into cryo-EM data. 著者: Linnea Yvonnesdotter / Urška Rovšnik / Christian Blau / Marie Lycksell / Rebecca Joy Howard / Erik Lindahl / 要旨: The resolution revolution has increasingly enabled single-particle cryogenic electron microscopy (cryo-EM) reconstructions of previously inaccessible systems, including membrane proteins-a category ...The resolution revolution has increasingly enabled single-particle cryogenic electron microscopy (cryo-EM) reconstructions of previously inaccessible systems, including membrane proteins-a category that constitutes a disproportionate share of drug targets. We present a protocol for using density-guided molecular dynamics simulations to automatically refine atomistic models into membrane protein cryo-EM maps. Using adaptive force density-guided simulations as implemented in the GROMACS molecular dynamics package, we show how automated model refinement of a membrane protein is achieved without the need to manually tune the fitting force ad hoc. We also present selection criteria to choose the best-fit model that balances stereochemistry and goodness of fit. The proposed protocol was used to refine models into a new cryo-EM density of the membrane protein maltoporin, either in a lipid bilayer or detergent micelle, and we found that results do not substantially differ from fitting in solution. Fitted structures satisfied classical model-quality metrics and improved the quality and the model-to-map correlation of the x-ray starting structure. Additionally, the density-guided fitting in combination with generalized orientation-dependent all-atom potential was used to correct the pixel-size estimation of the experimental cryo-EM density map. This work demonstrates the applicability of a straightforward automated approach to fitting membrane protein cryo-EM densities. Such computational approaches promise to facilitate rapid refinement of proteins under different conditions or with various ligands present, including targets in the highly relevant superfamily of membrane proteins. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 添付画像 |
|---|

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_15903.map.gz | 7.4 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-15903-v30.xmlemd-15903.xml | 16.2 KB 16.2 KB | 表示 表示 | EMDBヘッダ |
| FSC (解像度算出) | emd_15903_fsc.xml | 9.1 KB | 表示 | FSCデータファイル |
| 画像 | 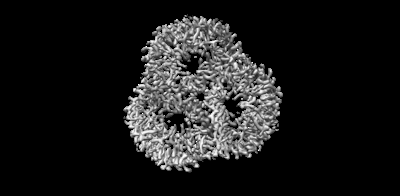 emd_15903.png emd_15903.png | 40.8 KB | ||
| マスクデータ | emd_15903_msk_1.map | 8 MB | マスクマップ | |
| Filedesc metadata | emd-15903.cif.gz | 5.2 KB | ||
| その他 | emd_15903_additional_1.map.gzemd_15903_half_map_1.map.gzemd_15903_half_map_2.map.gz | 3.4 MB 7.4 MB 7.4 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-15903ftp://ftp.pdbj.org/pub/emdb/structures/EMD-15903 http://ftp.pdbj.org/pub/emdb/structures/EMD-15903ftp://ftp.pdbj.org/pub/emdb/structures/EMD-15903 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_15903_validation.pdf.gz | 937.6 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_15903_full_validation.pdf.gz | 937.2 KB | 表示 | |
| XML形式データ | emd_15903_validation.xml.gz | 12.6 KB | 表示 | |
| CIF形式データ | emd_15903_validation.cif.gz | 17.1 KB | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-15903ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-15903 | HTTPS FTP |
-関連構造データ

-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_15903.map.gz / 形式: CCP4 / 大きさ: 8 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Half map 2, box 128 | ||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 0.83 Å | ||||||||||||||||||||||||||||||||||||
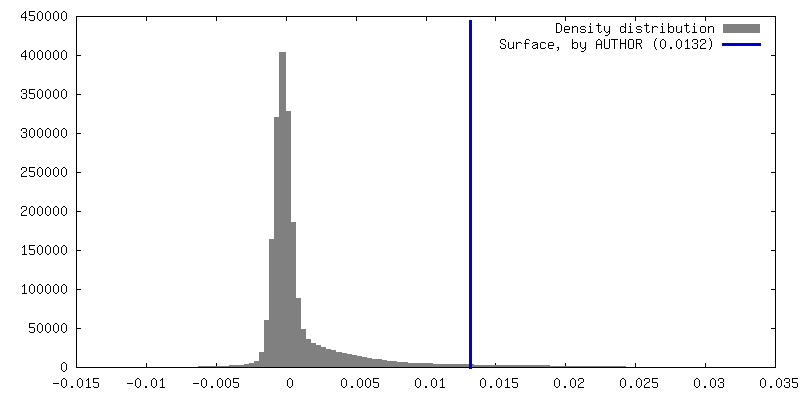
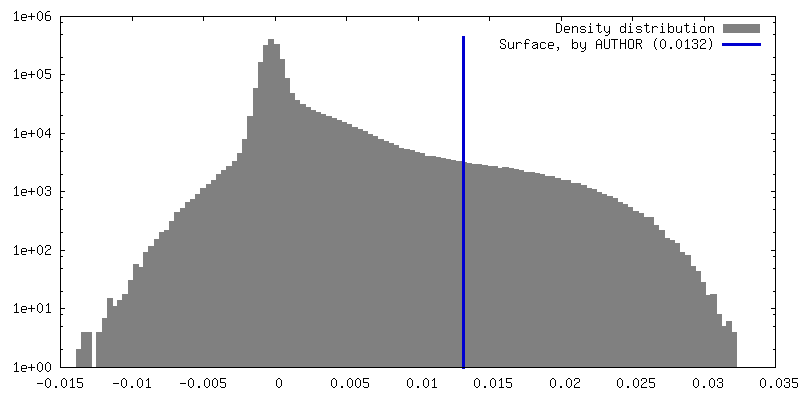
| 密度 |
| ||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)








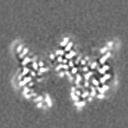


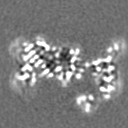
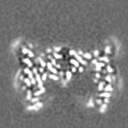

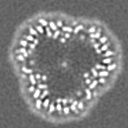

-添付データ
-マスク #1
| ファイル | emd_15903_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 投影像・断面図 |
| ||||||||||||
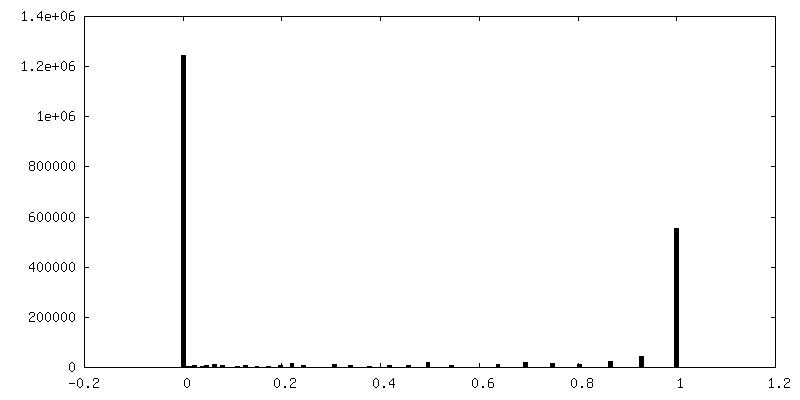
| 密度ヒストグラム |







-追加マップ: Postprocessed sharpened map, box 128
| ファイル | emd_15903_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Postprocessed sharpened map, box 128 | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
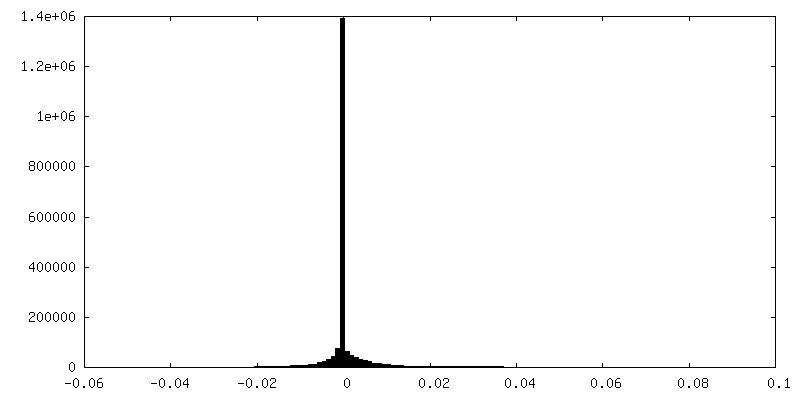
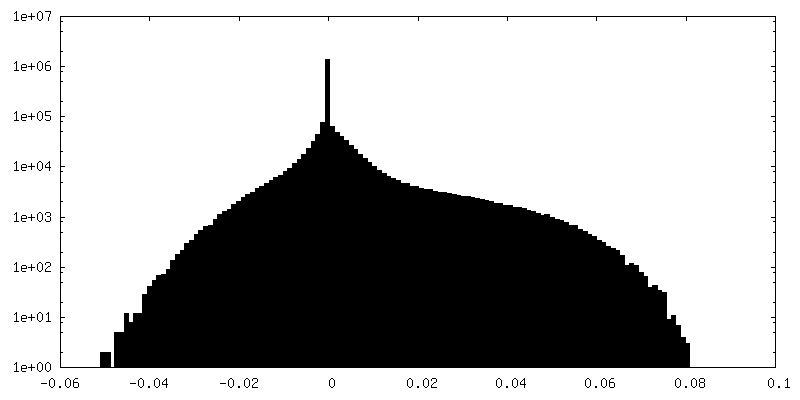
| 密度ヒストグラム |





-ハーフマップ: 3D refined model used for automated fitting, box 128
| ファイル | emd_15903_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | 3D refined model used for automated fitting, box 128 | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
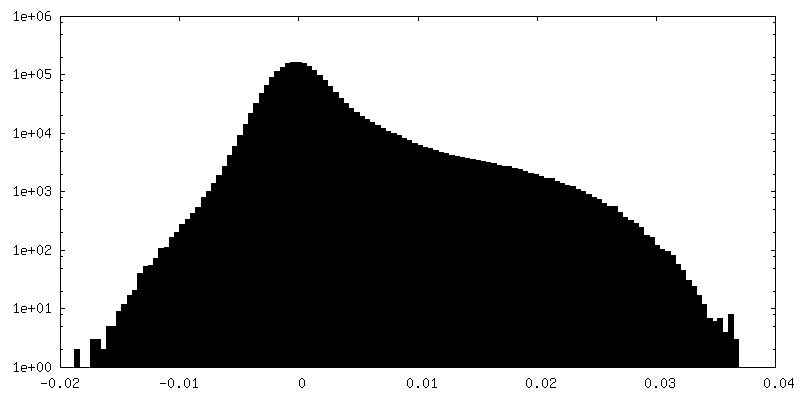
| 密度ヒストグラム |

-ハーフマップ: Half map 1, box 128
| ファイル | emd_15903_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Half map 1, box 128 | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |

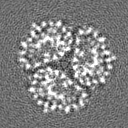
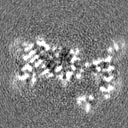
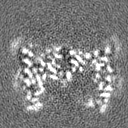
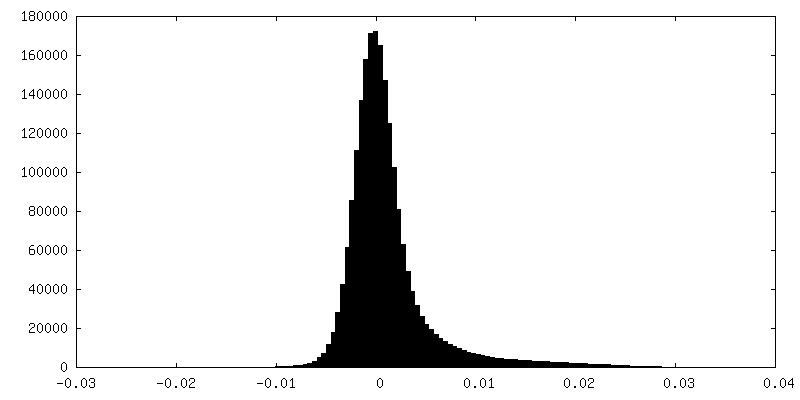

- 試料の構成要素
試料の構成要素
-全体 : Trimeric maltoporin (LamB protein)
| 全体 | 名称: Trimeric maltoporin (LamB protein) |
|---|---|
| 要素 |
|
-超分子 #1: Trimeric maltoporin (LamB protein)
| 超分子 | 名称: Trimeric maltoporin (LamB protein) / タイプ: complex / ID: 1 / 親要素: 0 / 含まれる分子: all |
|---|---|
| 由来(天然) | 生物種: |
-分子 #1: Maltoporin
| 分子 | 名称: Maltoporin / タイプ: protein_or_peptide / ID: 1 / コピー数: 3 / 光学異性体: LEVO |
|---|---|
| 由来(天然) | 生物種: |
| 分子量 | 理論値: 47.425785 KDa |
| 配列 | 文字列: VDFHGYARSG IGWTGSGGEQ QCFQTTGAQS KYRLGNECET YAELKLGQEV WKEGDKSFYF DTNVAYSVAQ QNDWEATDPA FREANVQGK NLIEWLPGST IWAGKRFYQR HDVHMIDFYY WDISGPGAGL ENIDVGFGKL SLAATRSSEA GGSSSFASNN I YDYTNETA ...文字列: VDFHGYARSG IGWTGSGGEQ QCFQTTGAQS KYRLGNECET YAELKLGQEV WKEGDKSFYF DTNVAYSVAQ QNDWEATDPA FREANVQGK NLIEWLPGST IWAGKRFYQR HDVHMIDFYY WDISGPGAGL ENIDVGFGKL SLAATRSSEA GGSSSFASNN I YDYTNETA NDVFDVRLAQ MEINPGGTLE LGVDYGRANL RDNYRLVDGA SKDGWLFTAE HTQSVLKGFN KFVVQYATDS MT SQGKGLS QGSGVAFDNE KFAYNINNNG HMLRILDHGA ISMGDNWDMM YVGMYQDINW DNDNGTKWWT VGIRPMYKWT PIM STVMEI GYDNVESQRT GDKNNQYKIT LAQQWQAGDS IWSRPAIRVF ATYAKWDEKW GYDYTGNADN NANFGKAVPA DFNG GSFGR GDSDEWTFGA QMEIWW UniProtKB: Maltoporin |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 3 mg/mL |
|---|---|
| 緩衝液 | pH: 7.4 |
| 凍結 | 凍結剤: ETHANE |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 撮影 | フィルム・検出器のモデル: GATAN K2 SUMMIT (4k x 4k) 検出モード: COUNTING / 平均電子線量: 42.0 e/Å2 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD 最大 デフォーカス(公称値): 2.8000000000000003 µm 最小 デフォーカス(公称値): 1.4000000000000001 µm |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |