Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Upright KimA dimer with bound c-di-AMP from B. subtilis | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | potassium importer / second messenger / c-di-AMP / MEMBRANE PROTEIN | |||||||||
| Function / homology | : / Amino acid/polyamine transporter I / Amino acid permease / symporter activity / potassium ion transport / metal ion binding / plasma membrane / Potassium transporter KimA Function and homology information Function and homology information | |||||||||
| Biological species |  | |||||||||
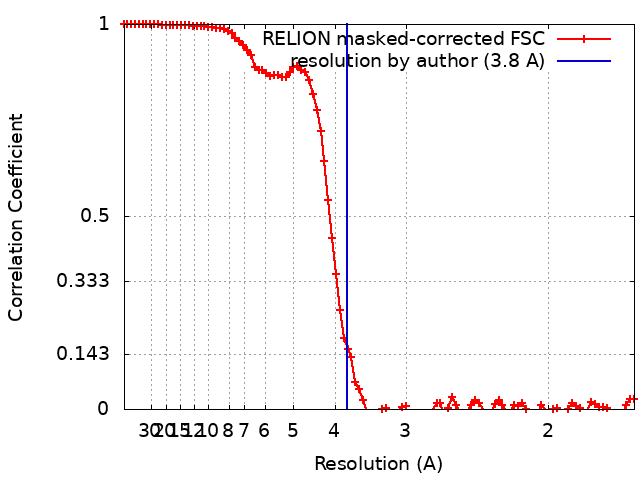
| Method | single particle reconstruction / cryo EM / Resolution: 3.8 Å | |||||||||
 Authors Authors | Vonck J / Wieferig JP | |||||||||
| Funding support |  Germany, 2 items Germany, 2 items
| |||||||||
 Citation Citation | Journal: Nat Commun / Year: 2023 Title: Cyclic di-AMP traps proton-coupled K transporters of the KUP family in an inward-occluded conformation. Authors: Michael F Fuss / Jan-Philip Wieferig / Robin A Corey / Yvonne Hellmich / Igor Tascón / Joana S Sousa / Phillip J Stansfeld / Janet Vonck / Inga Hänelt /   Abstract: Cyclic di-AMP is the only known essential second messenger in bacteria and archaea, regulating different proteins indispensable for numerous physiological processes. In particular, it controls ...Cyclic di-AMP is the only known essential second messenger in bacteria and archaea, regulating different proteins indispensable for numerous physiological processes. In particular, it controls various potassium and osmolyte transporters involved in osmoregulation. In Bacillus subtilis, the K/H symporter KimA of the KUP family is inactivated by c-di-AMP. KimA sustains survival at potassium limitation at low external pH by mediating potassium ion uptake. However, at elevated intracellular K concentrations, further K accumulation would be toxic. In this study, we reveal the molecular basis of how c-di-AMP binding inhibits KimA. We report cryo-EM structures of KimA with bound c-di-AMP in detergent solution and reconstituted in amphipols. By combining structural data with functional assays and molecular dynamics simulations we reveal how c-di-AMP modulates transport. We show that an intracellular loop in the transmembrane domain interacts with c-di-AMP bound to the adjacent cytosolic domain. This reduces the mobility of transmembrane helices at the cytosolic side of the K binding site and therefore traps KimA in an inward-occluded conformation. #1: Journal: Biorxiv / Year: 2023Title: Cyclic di-AMP traps proton-coupled K+ transporters of the KUP family in an inward-occluded conformation Authors: Fuss MF / Wieferig JP / Corey R / Hellmich Y / Tascon I / Sousa JS / Stansfeld P / Vonck J / Haenelt I | |||||||||
| History |
|
- Structure visualization
Structure visualization
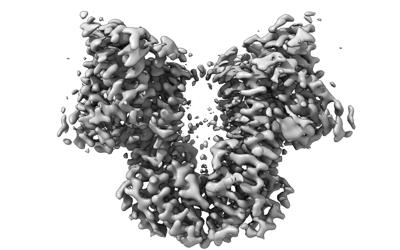
| Supplemental images |
|---|
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_15895.map.gz | 65.6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-15895-v30.xmlemd-15895.xml | 25.7 KB 25.7 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_15895_fsc.xml | 9.4 KB | Display | FSC data file |
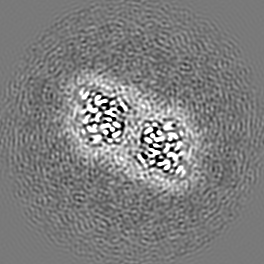
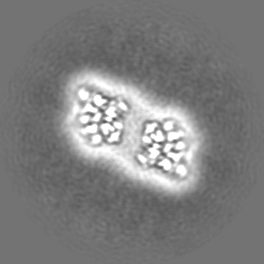
| Images | 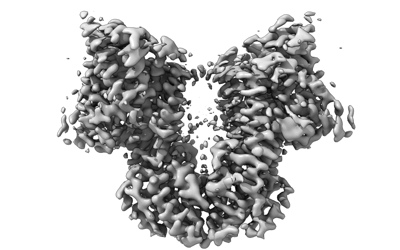 emd_15895.png emd_15895.png | 74.8 KB | ||
| Filedesc metadata | emd-15895.cif.gz | 6.6 KB | ||
| Others | emd_15895_additional_1.map.gzemd_15895_additional_2.map.gzemd_15895_additional_3.map.gzemd_15895_additional_4.map.gzemd_15895_half_map_1.map.gzemd_15895_half_map_2.map.gz | 52.9 MB 54.7 MB 54.7 MB 65.6 MB 53.4 MB 53.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-15895ftp://ftp.pdbj.org/pub/emdb/structures/EMD-15895 http://ftp.pdbj.org/pub/emdb/structures/EMD-15895ftp://ftp.pdbj.org/pub/emdb/structures/EMD-15895 | HTTPS FTP |
-Related structure data
| Related structure data |  8b71MC  8b70C M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |

-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_15895.map.gz / Format: CCP4 / Size: 70.2 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.831 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)

















-Supplemental data
-Additional map: unsharpened map
| File | emd_15895_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | unsharpened map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




-Additional map: Halfmap2 focused refinement half-dimer
| File | emd_15895_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Halfmap2 focused refinement half-dimer | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |


-Additional map: Halfmap1 focused refinement half-dimer
| File | emd_15895_additional_3.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Halfmap1 focused refinement half-dimer | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |

-Additional map: Focused refinement half-dimer
| File | emd_15895_additional_4.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Focused refinement half-dimer | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




-Half map: #2
| File | emd_15895_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |




-Half map: #1
| File | emd_15895_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |




- Sample components
Sample components
-Entire : Upright dimer KimA with c-di-AMP bound
| Entire | Name: Upright dimer KimA with c-di-AMP bound |
|---|---|
| Components |
|
-Supramolecule #1: Upright dimer KimA with c-di-AMP bound
| Supramolecule | Name: Upright dimer KimA with c-di-AMP bound / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 134 KDa |
-Macromolecule #1: Potassium transporter KimA
| Macromolecule | Name: Potassium transporter KimA / type: protein_or_peptide / ID: 1 / Number of copies: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 66.838977 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MYHSIKRFLI GKPLKSQAAG EQKLTKLKAL AMLSSDALSS VAYGTEQILI ILATISAAAF WYSIPIAVGV LILLLALILS YRQIIYAYP QGGGAYIVSK ENLGEKPGLI AGGSLLVDYI LTVAVSISAG TDAITSAFPA LHDYHVPIAI FLVLVIMILN L RGLSESAS ...String: MYHSIKRFLI GKPLKSQAAG EQKLTKLKAL AMLSSDALSS VAYGTEQILI ILATISAAAF WYSIPIAVGV LILLLALILS YRQIIYAYP QGGGAYIVSK ENLGEKPGLI AGGSLLVDYI LTVAVSISAG TDAITSAFPA LHDYHVPIAI FLVLVIMILN L RGLSESAS ILAYPVYLFV VALLVLIAVG LFKLMTGQID QPAHHTSLGT PVAGITLFLL LKAFSSGCSA LTGVEAISNA IP AFKNPPA RNAARTLAMM GILLAILFSG ITVLAYGYGT APKPDETVVS QIASETFGRN VFYYVIQGVT SLILVLAANT GFS AFPQLA FNLARDQYMP RMFTVRGDRL GFSNGIIFLG FASIVLIILF GGQTEHLIPL YAVGVFIPFT LSQTGMCMKW IKQK PKGWI GKMLINSCGA LISFMVLSIL FVTKFNVVWP VLIFMPIVVL LFFAIKNHYT AVGEQLRIVD KEPEEIKGTV VIVPV AGVT TVVQKSIHYA KSLSDQVIAV HVSFDREQEK KFEKRWEELN NGVRLVTLHS SYRSLVHPFD KFLETVEAKA KKEQFS VMV LFPQFITKKR WHTILHNQSA FLLRVRLFWK KDIMVATLPY HFKK UniProtKB: Potassium transporter KimA |
-Macromolecule #2: (2R,3R,3aS,5R,7aR,9R,10R,10aS,12R,14aR)-2,9-bis(6-amino-9H-purin-...
| Macromolecule | Name: (2R,3R,3aS,5R,7aR,9R,10R,10aS,12R,14aR)-2,9-bis(6-amino-9H-purin-9-yl)octahydro-2H,7H-difuro[3,2-d:3',2'-j][1,3,7,9,2,8 ]tetraoxadiphosphacyclododecine-3,5,10,12-tetrol 5,12-dioxide type: ligand / ID: 2 / Number of copies: 2 / Formula: 2BA |
|---|---|
| Molecular weight | Theoretical: 658.412 Da |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 2.25 mg/mL |
|---|---|
| Buffer | pH: 8 |
| Grid | Model: UltrAuFoil R1.2/1.3 / Material: GOLD / Mesh: 400 / Pretreatment - Type: GLOW DISCHARGE |
| Vitrification | Cryogen name: ETHANE |
| Details | Reconstituted in amphipols PMAL C8 |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Average electron dose: 55.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: SPOT SCAN / Imaging mode: BRIGHT FIELD / Nominal defocus max: 2.5 µm / Nominal defocus min: 1.1 µm |
| Sample stage | Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |