 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 |  | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | RQT-bound 80S ribosome from S. cerevisiae (C1, raw consensus map) | ||||||||||||
 マップデータ マップデータ | refined map of the RQTc1 ribsome | ||||||||||||
 試料 試料 |
| ||||||||||||
 キーワード キーワード | collision / RNA binding / RQT / RQC / RIBOSOME | ||||||||||||
| 生物種 |  | ||||||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 2.4 Å | ||||||||||||
 データ登録者 データ登録者 | Best KM / Ikeuchi K / Kater L / Best DM / Musial J / Matsuo Y / Berninghausen O / Becker T / Inada T / Beckmann R | ||||||||||||
| 資金援助 | European Union,  ドイツ, 3件 ドイツ, 3件
| ||||||||||||
 引用 引用 | ジャーナル: Acta Crystallogr D Struct Biol / 年: 2018 タイトル: Real-space refinement in PHENIX for cryo-EM and crystallography. 著者: Pavel V Afonine / Billy K Poon / Randy J Read / Oleg V Sobolev / Thomas C Terwilliger / Alexandre Urzhumtsev / Paul D Adams /    要旨: This article describes the implementation of real-space refinement in the phenix.real_space_refine program from the PHENIX suite. The use of a simplified refinement target function enables very fast ...This article describes the implementation of real-space refinement in the phenix.real_space_refine program from the PHENIX suite. The use of a simplified refinement target function enables very fast calculation, which in turn makes it possible to identify optimal data-restraint weights as part of routine refinements with little runtime cost. Refinement of atomic models against low-resolution data benefits from the inclusion of as much additional information as is available. In addition to standard restraints on covalent geometry, phenix.real_space_refine makes use of extra information such as secondary-structure and rotamer-specific restraints, as well as restraints or constraints on internal molecular symmetry. The re-refinement of 385 cryo-EM-derived models available in the Protein Data Bank at resolutions of 6 Å or better shows significant improvement of the models and of the fit of these models to the target maps. | ||||||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 添付画像 |
|---|

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_15228.map.gz | 334.9 MB |  EMDBマップデータ形式 EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-15228-v30.xmlemd-15228.xml | 37.2 KB 37.2 KB | 表示 表示 | EMDBヘッダ |
| FSC (解像度算出) | emd_15228_fsc.xml | 18.3 KB | 表示 | FSCデータファイル |
| 画像 | 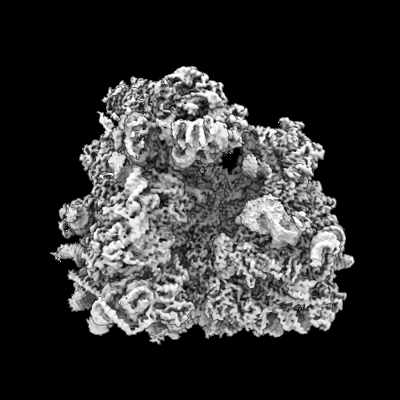 emd_15228.png emd_15228.png | 104.3 KB | ||
| その他 | emd_15228_half_map_1.map.gzemd_15228_half_map_2.map.gz | 622.4 MB 622.4 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-15228ftp://ftp.pdbj.org/pub/emdb/structures/EMD-15228 http://ftp.pdbj.org/pub/emdb/structures/EMD-15228ftp://ftp.pdbj.org/pub/emdb/structures/EMD-15228 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_15228_validation.pdf.gz | 954.7 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_15228_full_validation.pdf.gz | 954.3 KB | 表示 | |
| XML形式データ | emd_15228_validation.xml.gz | 28.3 KB | 表示 | |
| CIF形式データ | emd_15228_validation.cif.gz | 37.7 KB | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-15228ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-15228 | HTTPS FTP |
-関連構造データ
















-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |

-マップ
| ファイル | ダウンロード / ファイル: emd_15228.map.gz / 形式: CCP4 / 大きさ: 669.9 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | refined map of the RQTc1 ribsome | ||||||||||||||||||||||||||||||||||||
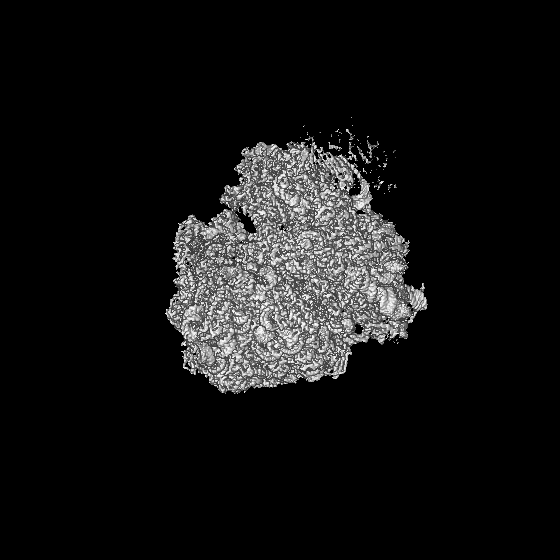
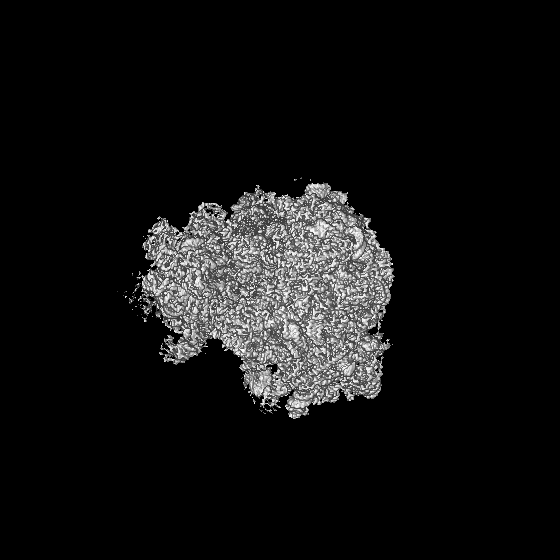
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.045 Å | ||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)

















-添付データ
-ハーフマップ: halfmap 1
| ファイル | emd_15228_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | halfmap 1 | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |







-ハーフマップ: halfmap 2
| ファイル | emd_15228_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | halfmap 2 | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |








- 試料の構成要素
試料の構成要素
-全体 : ribosome with bound RQT components (Slh1, Cue3 and Rqt4)
| 全体 | 名称: ribosome with bound RQT components (Slh1, Cue3 and Rqt4) |
|---|---|
| 要素 |
|
-超分子 #1: ribosome with bound RQT components (Slh1, Cue3 and Rqt4)
| 超分子 | 名称: ribosome with bound RQT components (Slh1, Cue3 and Rqt4) タイプ: complex / ID: 1 / 親要素: 0 / 含まれる分子: #1-#82 |
|---|
-超分子 #2: RQT complex (Slh1, Cue3 and Rqt4)
| 超分子 | 名称: RQT complex (Slh1, Cue3 and Rqt4) / タイプ: complex / ID: 2 / 親要素: 1 / 含まれる分子: #80-#82 |
|---|---|
| 由来(天然) | 生物種: |
-超分子 #3: ribosome
| 超分子 | 名称: ribosome / タイプ: complex / ID: 3 / 親要素: 1 / 含まれる分子: #1-#79 |
|---|---|
| 由来(天然) | 生物種: |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 緩衝液 | pH: 7.5 |
|---|---|
| グリッド | モデル: Quantifoil R3/3 / 材質: COPPER / 支持フィルム - 材質: CARBON |
| 凍結 | 凍結剤: ETHANE / 装置: FEI VITROBOT MARK II |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 撮影 | フィルム・検出器のモデル: GATAN K2 SUMMIT (4k x 4k) 検出モード: COUNTING / 平均電子線量: 43.6 e/Å2 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 照射モード: SPOT SCAN / 撮影モード: BRIGHT FIELD / 最大 デフォーカス(公称値): 3.0 µm / 最小 デフォーカス(公称値): 0.5 µm |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |

