German Federal Ministry for Education and Research
031L0100
Germany
Citation
Journal: Science / Year: 2022 Title: AI-based structure prediction empowers integrative structural analysis of human nuclear pores. Authors: Shyamal Mosalaganti / Agnieszka Obarska-Kosinska / Marc Siggel / Reiya Taniguchi / Beata Turoňová / Christian E Zimmerli / Katarzyna Buczak / Florian H Schmidt / Erica Margiotta / Marie- ...Authors: Shyamal Mosalaganti / Agnieszka Obarska-Kosinska / Marc Siggel / Reiya Taniguchi / Beata Turoňová / Christian E Zimmerli / Katarzyna Buczak / Florian H Schmidt / Erica Margiotta / Marie-Therese Mackmull / Wim J H Hagen / Gerhard Hummer / Jan Kosinski / Martin Beck / Abstract: INTRODUCTION The eukaryotic nucleus pro-tects the genome and is enclosed by the two membranes of the nuclear envelope. Nuclear pore complexes (NPCs) perforate the nuclear envelope to facilitate ...INTRODUCTION The eukaryotic nucleus pro-tects the genome and is enclosed by the two membranes of the nuclear envelope. Nuclear pore complexes (NPCs) perforate the nuclear envelope to facilitate nucleocytoplasmic transport. With a molecular weight of ∼120 MDa, the human NPC is one of the larg-est protein complexes. Its ~1000 proteins are taken in multiple copies from a set of about 30 distinct nucleoporins (NUPs). They can be roughly categorized into two classes. Scaf-fold NUPs contain folded domains and form a cylindrical scaffold architecture around a central channel. Intrinsically disordered NUPs line the scaffold and extend into the central channel, where they interact with cargo complexes. The NPC architecture is highly dynamic. It responds to changes in nuclear envelope tension with conforma-tional breathing that manifests in dilation and constriction movements. Elucidating the scaffold architecture, ultimately at atomic resolution, will be important for gaining a more precise understanding of NPC function and dynamics but imposes a substantial chal-lenge for structural biologists. RATIONALE Considerable progress has been made toward this goal by a joint effort in the field. A synergistic combination of complementary approaches has turned out to be critical. In situ structural biology techniques were used to reveal the overall layout of the NPC scaffold that defines the spatial reference for molecular modeling. High-resolution structures of many NUPs were determined in vitro. Proteomic analysis and extensive biochemical work unraveled the interaction network of NUPs. Integra-tive modeling has been used to combine the different types of data, resulting in a rough outline of the NPC scaffold. Previous struc-tural models of the human NPC, however, were patchy and limited in accuracy owing to several challenges: (i) Many of the high-resolution structures of individual NUPs have been solved from distantly related species and, consequently, do not comprehensively cover their human counterparts. (ii) The scaf-fold is interconnected by a set of intrinsically disordered linker NUPs that are not straight-forwardly accessible to common structural biology techniques. (iii) The NPC scaffold intimately embraces the fused inner and outer nuclear membranes in a distinctive topol-ogy and cannot be studied in isolation. (iv) The conformational dynamics of scaffold NUPs limits the resolution achievable in structure determination. RESULTS In this study, we used artificial intelligence (AI)-based prediction to generate an exten-sive repertoire of structural models of human NUPs and their subcomplexes. The resulting models cover various domains and interfaces that so far remained structurally uncharac-terized. Benchmarking against previous and unpublished x-ray and cryo-electron micros-copy structures revealed unprecedented accu-racy. We obtained well-resolved cryo-electron tomographic maps of both the constricted and dilated conformational states of the hu-man NPC. Using integrative modeling, we fit-ted the structural models of individual NUPs into the cryo-electron microscopy maps. We explicitly included several linker NUPs and traced their trajectory through the NPC scaf-fold. We elucidated in great detail how mem-brane-associated and transmembrane NUPs are distributed across the fusion topology of both nuclear membranes. The resulting architectural model increases the structural coverage of the human NPC scaffold by about twofold. We extensively validated our model against both earlier and new experimental data. The completeness of our model has enabled microsecond-long coarse-grained molecular dynamics simulations of the NPC scaffold within an explicit membrane en-vironment and solvent. These simulations reveal that the NPC scaffold prevents the constriction of the otherwise stable double-membrane fusion pore to small diameters in the absence of membrane tension. CONCLUSION Our 70-MDa atomically re-solved model covers >90% of the human NPC scaffold. It captures conforma-tional changes that occur during dilation and constriction. It also reveals the precise anchoring sites for intrinsically disordered NUPs, the identification of which is a prerequisite for a complete and dy-namic model of the NPC. Our study exempli-fies how AI-based structure prediction may accelerate the elucidation of subcellular ar-chitecture at atomic resolution. [Figure: see text].
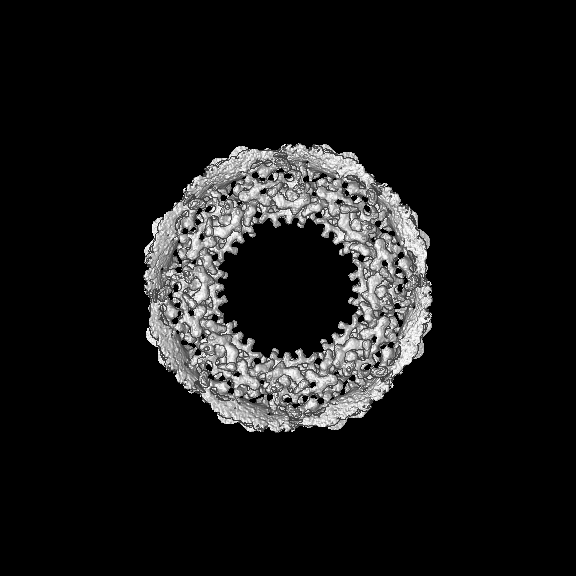
Entire : Cytoplasmic ring of the human nuclear pore complex
Entire
Name: Cytoplasmic ring of the human nuclear pore complex
Components
Organelle or cellular component: Cytoplasmic ring of the human nuclear pore complex
-
Supramolecule #1: Cytoplasmic ring of the human nuclear pore complex
Supramolecule
Name: Cytoplasmic ring of the human nuclear pore complex / type: organelle_or_cellular_component / ID: 1 / Parent: 0 / Macromolecule list: #1-#25
Source (natural)
Organism: Homo sapiens (human)
Molecular weight
Theoretical: 50 MDa
-
Experimental details
-
Structure determination
Method
cryo EM
Processing
subtomogram averaging
Aggregation state
cell
-
Sample preparation
Buffer
pH: 7.5
Vitrification
Cryogen name: ETHANE / Instrument: LEICA EM GP
-
Electron microscopy
Microscope
TFS KRIOS
Image recording
Film or detector model: GATAN K2 QUANTUM (4k x 4k) / Average electron dose: 2.93 e/Å2
Electron beam
Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN
Electron optics
C2 aperture diameter: 70.0 µm / Illumination mode: OTHER / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 4.0 µm / Nominal defocus min: 2.0 µm
Experimental equipment
Model: Titan Krios / Image courtesy: FEI Company
-
Image processing
Final reconstruction
Applied symmetry - Point group: C1 (asymmetric) / Resolution.type: BY AUTHOR / Resolution: 12.6 Å / Resolution method: FSC 0.143 CUT-OFF / Number subtomograms used: 30000
Extraction
Number tomograms: 554 / Number images used: 7711 Details: Full pores were extracted, which were subsequently split into asymmetric units (61688 subtomograms).
Final angle assignment
Type: OTHER
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information
 Map data
Map data Sample
Sample Homo sapiens (human)
Homo sapiens (human) Authors
Authors Germany, 2 items
Germany, 2 items  Citation
Citation
 Structure visualization
Structure visualization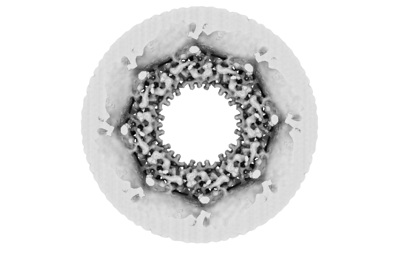
 Downloads & links
Downloads & links EMDB map data format
EMDB map data format emd_14328.png
emd_14328.png http://ftp.pdbj.org/pub/emdb/structures/EMD-14328
http://ftp.pdbj.org/pub/emdb/structures/EMD-14328









 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)


















 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN
