 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-12316 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | D1-state of wild type human mitochondrial LONP1 protease | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | human mitochondrial AAA+ protease / MOTOR PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationoxidation-dependent protein catabolic process / response to aluminum ion / PH domain binding / endopeptidase La / mitochondrial protein catabolic process / G-quadruplex DNA binding / ATP-dependent peptidase activity / protein quality control for misfolded or incompletely synthesized proteins / mitochondrial nucleoid / insulin receptor substrate binding ...oxidation-dependent protein catabolic process / response to aluminum ion / PH domain binding / endopeptidase La / mitochondrial protein catabolic process / G-quadruplex DNA binding / ATP-dependent peptidase activity / protein quality control for misfolded or incompletely synthesized proteins / mitochondrial nucleoid / insulin receptor substrate binding / Mitochondrial unfolded protein response (UPRmt) / chaperone-mediated protein complex assembly / DNA polymerase binding / response to hormone / negative regulation of insulin receptor signaling pathway / Mitochondrial protein degradation / : / mitochondrion organization / ADP binding / single-stranded DNA binding / cellular response to oxidative stress / sequence-specific DNA binding / response to hypoxia / single-stranded RNA binding / mitochondrial matrix / serine-type endopeptidase activity / ATP hydrolysis activity / mitochondrion / nucleoplasm / ATP binding / membrane / identical protein binding / cytosol Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 15.0 Å | |||||||||
 Authors Authors | Mohammed I / Schmitz KA | |||||||||
 Citation Citation | Journal: Structure / Year: 2022 Title: Catalytic cycling of human mitochondrial Lon protease. Authors: Inayathulla Mohammed / Kai A Schmitz / Niko Schenck / Dimitrios Balasopoulos / Annika Topitsch / Timm Maier / Jan Pieter Abrahams /  Abstract: The mitochondrial Lon protease (LonP1) regulates mitochondrial health by removing redundant proteins from the mitochondrial matrix. We determined LonP1 in eight nucleotide-dependent conformational ...The mitochondrial Lon protease (LonP1) regulates mitochondrial health by removing redundant proteins from the mitochondrial matrix. We determined LonP1 in eight nucleotide-dependent conformational states by cryoelectron microscopy (cryo-EM). The flexible assembly of N-terminal domains had 3-fold symmetry, and its orientation depended on the conformational state. We show that a conserved structural motif around T803 with a high similarity to the trypsin catalytic triad is essential for proteolysis. We show that LonP1 is not regulated by redox potential, despite the presence of two conserved cysteines at disulfide-bonding distance in its unfoldase core. Our data indicate how sequential ATP hydrolysis controls substrate protein translocation in a 6-fold binding change mechanism. Substrate protein translocation, rather than ATP hydrolysis, is a rate-limiting step, suggesting that LonP1 is a Brownian ratchet with ATP hydrolysis preventing translocation reversal. 3-fold rocking motions of the flexible N-domain assembly may assist thermal unfolding of the substrate protein. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_12316.map.gz | 59.8 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-12316-v30.xmlemd-12316.xml | 19.7 KB 19.7 KB | Display Display | EMDB header |
| Images | 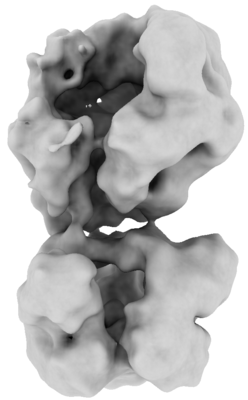 emd_12316.png emd_12316.png | 52.3 KB | ||
| Filedesc metadata | emd-12316.cif.gz | 6.8 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-12316ftp://ftp.pdbj.org/pub/emdb/structures/EMD-12316 http://ftp.pdbj.org/pub/emdb/structures/EMD-12316ftp://ftp.pdbj.org/pub/emdb/structures/EMD-12316 | HTTPS FTP |
-Related structure data
| Related structure data |  7ngpMC  7nfyC  7ng4C  7ng5C  7ngcC  7ngfC  7nglC  7ngqC  7oxoC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |














-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_12316.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.6531 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
- Sample components
Sample components
-Entire : D1-state of LonP1 hexameric complex
| Entire | Name: D1-state of LonP1 hexameric complex |
|---|---|
| Components |
|
-Supramolecule #1: D1-state of LonP1 hexameric complex
| Supramolecule | Name: D1-state of LonP1 hexameric complex / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 600 KDa |
-Macromolecule #1: Lon protease homolog, mitochondrial
| Macromolecule | Name: Lon protease homolog, mitochondrial / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO / EC number: endopeptidase La |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 93.201383 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: HLPLIAITRN PVFPRFIKII EVKNKKLVEL LRRKVRLAQP YVGVFLKRDD SNESDVVESL DEIYHTGTFA QIHEMQDLGD KLRMIVMGH RRVHISRQLE VEPEEPEAEN KHKPRRKSKR GKKEAEDELS ARHPAELAME PTPELPAEVL MVEVENVVHE D FQVTEEVK ...String: HLPLIAITRN PVFPRFIKII EVKNKKLVEL LRRKVRLAQP YVGVFLKRDD SNESDVVESL DEIYHTGTFA QIHEMQDLGD KLRMIVMGH RRVHISRQLE VEPEEPEAEN KHKPRRKSKR GKKEAEDELS ARHPAELAME PTPELPAEVL MVEVENVVHE D FQVTEEVK ALTAEIVKTI RDIIALNPLY RESVLQMMQA GQRVVDNPIY LSDMGAALTG AESHELQDVL EETNIPKRLY KA LSLLKKE FELSKLQQRL GREVEEKIKQ THRKYLLQEQ LKIIKKELGL EKDDKDAIEE KFRERLKELV VPKHVMDVVD EEL SKLGLL DNHSSEFNVT RNYLDWLTSI PWGKYSNENL DLARAQAVLE EDHYGMEDVK KRILEFIAVS QLRGSTQGKI LCFY GPPGV GKTSIARSIA RALNREYFRF SVGGMTDVAE IKGHRRTYVG AMPGKIIQCL KKTKTENPLI LIDEVDKIGR GYQGD PSSA LLELLDPEQN ANFLDHYLDV PVDLSKVLFI CTANVTDTIP EPLRDRMEMI NVSGYVAQEK LAIAERYLVP QARALC GLD ESKAKLSSDV LTLLIKQYCR ESGVRNLQKQ VEKVLRKSAY KIVSGEAESV EVTPENLQDF VGKPVFTVER MYDVTPP GV VMGLAWTAMG GSTLFVETSL RRPQDKDAKG DKDGSLEVTG QLGEVMKESA RIAYTFARAF LMQHAPANDY LVTSHIHL H VPEGATPKDG PSAGCTIVTA LLSLAMGRPV RQNLAMTGEV SLTGKILPVG GIKEKTIAAK RAGVTCIVLP AENKKDFYD LAAFITEGLE VHFVEHYREI FDIAFP UniProtKB: Lon protease homolog, mitochondrial |
-Macromolecule #2: ADENOSINE-5'-DIPHOSPHATE
| Macromolecule | Name: ADENOSINE-5'-DIPHOSPHATE / type: ligand / ID: 2 / Number of copies: 6 / Formula: ADP |
|---|---|
| Molecular weight | Theoretical: 427.201 Da |
| Chemical component information |  ChemComp-ADP: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.45 mg/mL |
|---|---|
| Buffer | pH: 7.4 |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Average electron dose: 64.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: OTHER / Imaging mode: BRIGHT FIELD |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Refinement | Protocol: RIGID BODY FIT |
|---|---|
| Output model | PDB-7ngp: |
