 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-12047: CryoEM Structure of the yeast peroxisomal membrane Pex14p/Pex17p ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-12047 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
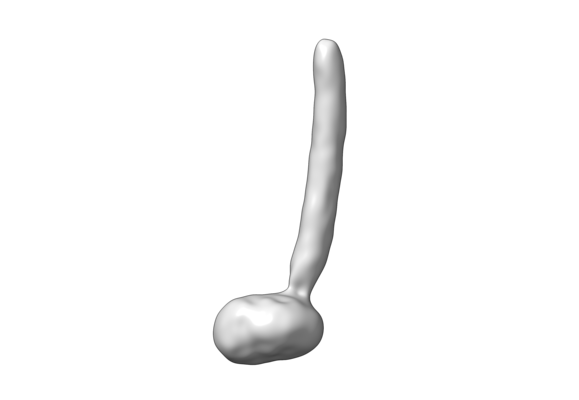
| Title | CryoEM Structure of the yeast peroxisomal membrane Pex14p/Pex17p complex | ||||||||||||
 Map data Map data | CryoEM Structure of the Pex14p/Pex17p complex | ||||||||||||
 Sample Sample |
| ||||||||||||
| Biological species |  | ||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 10.2 Å | ||||||||||||
 Authors Authors | Lill P / Gatsogiannis C | ||||||||||||
| Funding support |  Germany, 3 items Germany, 3 items
| ||||||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2020 Title: Towards the molecular architecture of the peroxisomal receptor docking complex. Authors: Pascal Lill / Tobias Hansen / Daniel Wendscheck / Bjoern Udo Klink / Tomasz Jeziorek / Dimitrios Vismpas / Jonas Miehling / Julian Bender / Andreas Schummer / Friedel Drepper / Wolfgang ...Authors: Pascal Lill / Tobias Hansen / Daniel Wendscheck / Bjoern Udo Klink / Tomasz Jeziorek / Dimitrios Vismpas / Jonas Miehling / Julian Bender / Andreas Schummer / Friedel Drepper / Wolfgang Girzalsky / Bettina Warscheid / Ralf Erdmann / Christos Gatsogiannis / Abstract: Import of yeast peroxisomal matrix proteins is initiated by cytosolic receptors, which specifically recognize and bind the respective cargo proteins. At the peroxisomal membrane, the cargo-loaded ...Import of yeast peroxisomal matrix proteins is initiated by cytosolic receptors, which specifically recognize and bind the respective cargo proteins. At the peroxisomal membrane, the cargo-loaded receptor interacts with the docking protein Pex14p that is tightly associated with Pex17p. Previous data suggest that this interaction triggers the formation of an import pore for further translocation of the cargo. The mechanistic principles, however, are unclear, mainly because structures of higher-order assemblies are still lacking. Here, using an integrative approach, we provide the structural characterization of the major components of the peroxisomal docking complex Pex14p/Pex17p, in a native bilayer environment, and reveal its subunit organization. Our data show that three copies of Pex14p and a single copy of Pex17p assemble to form a 20-nm rod-like particle. The different subunits are arranged in a parallel manner, showing interactions along their complete sequences and providing receptor binding sites on both membrane sides. The long rod facing the cytosol is mainly formed by the predicted coiled-coil domains of Pex14p and Pex17p, possibly providing the necessary structural support for the formation of the import pore. Further implications of Pex14p/Pex17p for formation of the peroxisomal translocon are discussed. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_12047.map.gz | 6.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-12047-v30.xmlemd-12047.xml | 17 KB 17 KB | Display Display | EMDB header |
| Images |  emd_12047.png emd_12047.png | 21.5 KB | ||
| Others | emd_12047_additional_1.map.gz | 3.6 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-12047ftp://ftp.pdbj.org/pub/emdb/structures/EMD-12047 http://ftp.pdbj.org/pub/emdb/structures/EMD-12047ftp://ftp.pdbj.org/pub/emdb/structures/EMD-12047 | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_12047.map.gz / Format: CCP4 / Size: 103 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | CryoEM Structure of the Pex14p/Pex17p complex | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.09 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Additional map: CryoEM structure of the rod domain of the...
| File | emd_12047_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | CryoEM structure of the rod domain of the Pex14p/Pex17p complex upon signal subtraction of the nanodisc density and subsequent refinement. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






- Sample components
Sample components
-Entire : Peroxisomal Membrane Complex Pex14p/Pex17p reconstituted in cMSP1...
| Entire | Name: Peroxisomal Membrane Complex Pex14p/Pex17p reconstituted in cMSP1D1dH4-6 nanodiscs |
|---|---|
| Components |
|
-Supramolecule #1: Peroxisomal Membrane Complex Pex14p/Pex17p reconstituted in cMSP1...
| Supramolecule | Name: Peroxisomal Membrane Complex Pex14p/Pex17p reconstituted in cMSP1D1dH4-6 nanodiscs type: complex / ID: 1 / Parent: 0 / Macromolecule list: all Details: Pex14p and Pex17p constitute the major components of the peroxisomal membrane docking complex. Pex14p and Pex17p form a 3:1 heterotetrameric complex. |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 144 KDa |
-Macromolecule #1: Peroxisomal membrane protein Pex14p
| Macromolecule | Name: Peroxisomal membrane protein Pex14p / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism:  |
| Sequence | String: MSDVVSKDRK ALFDSAVSFL KDESIKDAPL LKKIEFLKSK GLTEKEIEIA MKEPKKDGIV GDEVSKKIGS TENRASQDM YLYEAMPPTL PHRDWKDYFV MATATAGLLY GAYEVTRRYV IPNILPEAKS KLEGDKKEID D QFSKIDTV LNAIEAEQAE FRKKESETLK ...String: MSDVVSKDRK ALFDSAVSFL KDESIKDAPL LKKIEFLKSK GLTEKEIEIA MKEPKKDGIV GDEVSKKIGS TENRASQDM YLYEAMPPTL PHRDWKDYFV MATATAGLLY GAYEVTRRYV IPNILPEAKS KLEGDKKEID D QFSKIDTV LNAIEAEQAE FRKKESETLK ELSDTIAELK QALVQTTRSR EKIEDEFRIV KLEVVNMQNT ID KFVSDND GMQELNNIQK EMESLKSLMN NRMESGNAQD NRLFSISPNG IPGIDTIPSA SEILAKMGMQ EES DKEKEN GSDANKDDNA VPAWKKAREQ TIDSNASIPE WQKNTAANEI SVPDWQNGQV EDSIP |
-Macromolecule #2: Peroxisomal membrane protein Pex17p
| Macromolecule | Name: Peroxisomal membrane protein Pex17p / type: protein_or_peptide / ID: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism: |
| Sequence | String: MTSINSFPRN IDWPSNIGIK KIEGTNPTVN AIKGLLYNGG SIYAFLYFVI AMFVEPTLQK QYQQRNDFSL FVLLRLRRI IAQLQKRLVM TPVSSLGFNE QNNFVERSTQ TSDDNIIRED NSHWAEMIYQ LQNMKQELQY F NRSSGQPS ESIDDFVFQI KMVTDQVELT ...String: MTSINSFPRN IDWPSNIGIK KIEGTNPTVN AIKGLLYNGG SIYAFLYFVI AMFVEPTLQK QYQQRNDFSL FVLLRLRRI IAQLQKRLVM TPVSSLGFNE QNNFVERSTQ TSDDNIIRED NSHWAEMIYQ LQNMKQELQY F NRSSGQPS ESIDDFVFQI KMVTDQVELT DRSRAFSNKS RNIIQGIREI KGWFVNGQVP R |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.15 mg/mL | ||||||
|---|---|---|---|---|---|---|---|
| Buffer | pH: 8 / Component:
| ||||||
| Grid | Model: Quantifoil / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE | ||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK II | ||||||
| Details | The complex was reconstituted in circularMSP1D1D4-6 nanodiscs. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Phase plate: VOLTA PHASE PLATE |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Frames/image: 1-40 / Number grids imaged: 1 / Number real images: 3282 / Average exposure time: 15.0 sec. / Average electron dose: 56.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 0.0 mm / Nominal magnification: 130000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
