 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-11200: S-shaped FtsH dodecamer of A. aeolicus with lamellar-like N-termi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-11200 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
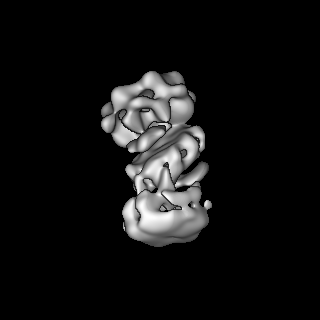
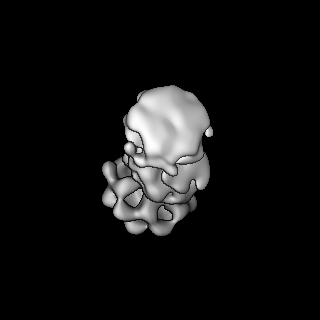
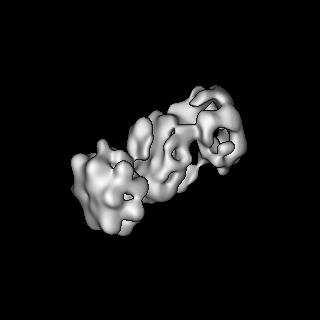
| Title | S-shaped FtsH dodecamer of A. aeolicus with lamellar-like N-terminal domains in LMNG micelle | |||||||||
 Map data Map data | S-shaped FtsH dodecamer of A. aeolicus with lamellar-like N-terminal domains in LMNG micelle | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  | |||||||||
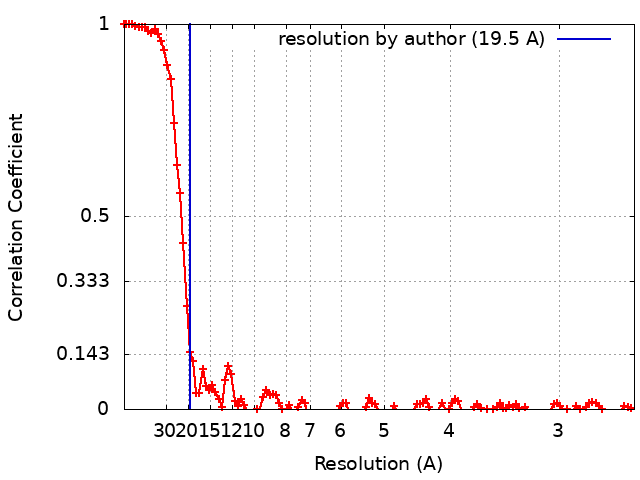
| Method | single particle reconstruction / cryo EM / Resolution: 19.5 Å | |||||||||
 Authors Authors | Carvalho V / Prabudiansyah I / Kovacik L / Chami M / Kieffer R / van der Valk R / de Lange N / Engel A / Aubin-Tam M-E | |||||||||
| Funding support |  Netherlands, Netherlands,  Switzerland, 2 items Switzerland, 2 items
| |||||||||
 Citation Citation | Journal: J Biol Chem / Year: 2021 Title: The cytoplasmic domain of the AAA+ protease FtsH is tilted with respect to the membrane to facilitate substrate entry. Authors: Vanessa Carvalho / Irfan Prabudiansyah / Lubomir Kovacik / Mohamed Chami / Roland Kieffer / Ramon van der Valk / Nick de Lange / Andreas Engel / Marie-Eve Aubin-Tam / Abstract: AAA+ proteases are degradation machines that use ATP hydrolysis to unfold protein substrates and translocate them through a central pore toward a degradation chamber. FtsH, a bacterial membrane- ...AAA+ proteases are degradation machines that use ATP hydrolysis to unfold protein substrates and translocate them through a central pore toward a degradation chamber. FtsH, a bacterial membrane-anchored AAA+ protease, plays a vital role in membrane protein quality control. How substrates reach the FtsH central pore is an open key question that is not resolved by the available atomic structures of cytoplasmic and periplasmic domains. In this work, we used both negative stain TEM and cryo-EM to determine 3D maps of the full-length Aquifex aeolicus FtsH protease. Unexpectedly, we observed that detergent solubilization induces the formation of fully active FtsH dodecamers, which consist of two FtsH hexamers in a single detergent micelle. The striking tilted conformation of the cytosolic domain in the FtsH dodecamer visualized by negative stain TEM suggests a lateral substrate entrance between the membrane and cytosolic domain. Such a substrate path was then resolved in the cryo-EM structure of the FtsH hexamer. By mapping the available structural information and structure predictions for the transmembrane helices to the amino acid sequence we identified a linker of ∼20 residues between the second transmembrane helix and the cytosolic domain. This unique polypeptide appears to be highly flexible and turned out to be essential for proper functioning of FtsH as its deletion fully eliminated the proteolytic activity of FtsH. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_11200.map.gz | 9.6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-11200-v30.xmlemd-11200.xml | 16.8 KB 16.8 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_11200_fsc.xml | 11.3 KB | Display | FSC data file |
| Images |  emd_11200.png emd_11200.png | 40.8 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-11200ftp://ftp.pdbj.org/pub/emdb/structures/EMD-11200 http://ftp.pdbj.org/pub/emdb/structures/EMD-11200ftp://ftp.pdbj.org/pub/emdb/structures/EMD-11200 | HTTPS FTP |
-Related structure data
| Related structure data | C: citing same article ( |
|---|---|
| Similar structure data |











-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_11200.map.gz / Format: CCP4 / Size: 125 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | S-shaped FtsH dodecamer of A. aeolicus with lamellar-like N-terminal domains in LMNG micelle | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.28 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)
















-Supplemental data
- Sample components
Sample components
-Entire : FtsH protease of A. aeolicus
| Entire | Name: FtsH protease of A. aeolicus |
|---|---|
| Components |
|
-Supramolecule #1: FtsH protease of A. aeolicus
| Supramolecule | Name: FtsH protease of A. aeolicus / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism: |
| Molecular weight | Experimental: 427 KDa |
-Macromolecule #1: A. aeolicus FtsH protease
| Macromolecule | Name: A. aeolicus FtsH protease / type: other / ID: 1 / Classification: other |
|---|---|
| Source (natural) | Organism: |
| Sequence | String: MNALKNFFIW AIIIGAAIVA FNLFEGKREF TTKVSLNEVV KLVEEGKVSY AEVRGNTAII QTKDGQKLE VTLPPNTNLV DKMVEKGVRV EVANPEPPGG WLVNVFLSWL PILFFIGIWI F LLRQMSGG GNVNRAFNFG KSRAKVYIEE KPKVTFKDVA GIEEVKEEVK ...String: MNALKNFFIW AIIIGAAIVA FNLFEGKREF TTKVSLNEVV KLVEEGKVSY AEVRGNTAII QTKDGQKLE VTLPPNTNLV DKMVEKGVRV EVANPEPPGG WLVNVFLSWL PILFFIGIWI F LLRQMSGG GNVNRAFNFG KSRAKVYIEE KPKVTFKDVA GIEEVKEEVK EIIEYLKDPV KF QKLGGRP PKGVLLYGEP GVGKTLLAKA IAGEAHVPFI SVSGSDFVEM FVGVGAARVR DLF ETAKKH APCIIFIDEI DAVGRARGAI PVGGGHDERE QTLNQLLVEM DGFDTSDGII VIAA TNRPD ILDPALLRPG RFDRQIFIPK PDVRGRYEIL KVHARNKKLA KDVDLEFVAR ATPGF TGAD LENLLNEAAL LAARKGKEEI TMEEIEEALD RITMGLERKG MTISPKEKEK IAIHEA GHA LMGLVSDDDD KVHKISIIPR GMALGVTQQL PIEDKHIYDK KDLYNKILVL LGGRAAE EV FFGKDGITTG AENDLQRATD LAYRMVSMWG MSDKVGPIAI RRVANPFLGG MTTAVDTS P DLLREIDEEV KRIITEQYEK AKAIVEEYKE PLKAVVKKLL EKETITCEEF VEVFKLYGI ELKDKCKKEE LFDKDRKSEE NKELKSEEVK EEVV |
| Recombinant expression | Organism: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.7 mg/mL | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 8 Component:
| |||||||||||||||
| Grid | Model: Quantifoil / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: AIR / Pretreatment - Pressure: 0.005 kPa | |||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 90.0 K / Max: 100.0 K |
| Specialist optics | Energy filter - Name: GIF Quantum LS / Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3838 pixel / Digitization - Dimensions - Height: 3760 pixel / Digitization - Sampling interval: 5.0 µm / Digitization - Frames/image: 1-35 / Number grids imaged: 1 / Number real images: 3993 / Average exposure time: 16.0 sec. / Average electron dose: 53.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Calibrated defocus max: 2.5 µm / Calibrated defocus min: 0.7000000000000001 µm / Calibrated magnification: 78247 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 0.7000000000000001 µm / Nominal magnification: 215000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Chain ID: A |
|---|---|
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: correlation coefficient |

