Netherlands Organisation for Scientific Research (NWO)
723-016-007
オランダ
Swiss National Science Foundation
18272.1
スイス
引用
ジャーナル: J Biol Chem / 年: 2021 タイトル: The cytoplasmic domain of the AAA+ protease FtsH is tilted with respect to the membrane to facilitate substrate entry. 著者: Vanessa Carvalho / Irfan Prabudiansyah / Lubomir Kovacik / Mohamed Chami / Roland Kieffer / Ramon van der Valk / Nick de Lange / Andreas Engel / Marie-Eve Aubin-Tam / 要旨: AAA+ proteases are degradation machines that use ATP hydrolysis to unfold protein substrates and translocate them through a central pore toward a degradation chamber. FtsH, a bacterial membrane- ...AAA+ proteases are degradation machines that use ATP hydrolysis to unfold protein substrates and translocate them through a central pore toward a degradation chamber. FtsH, a bacterial membrane-anchored AAA+ protease, plays a vital role in membrane protein quality control. How substrates reach the FtsH central pore is an open key question that is not resolved by the available atomic structures of cytoplasmic and periplasmic domains. In this work, we used both negative stain TEM and cryo-EM to determine 3D maps of the full-length Aquifex aeolicus FtsH protease. Unexpectedly, we observed that detergent solubilization induces the formation of fully active FtsH dodecamers, which consist of two FtsH hexamers in a single detergent micelle. The striking tilted conformation of the cytosolic domain in the FtsH dodecamer visualized by negative stain TEM suggests a lateral substrate entrance between the membrane and cytosolic domain. Such a substrate path was then resolved in the cryo-EM structure of the FtsH hexamer. By mapping the available structural information and structure predictions for the transmembrane helices to the amino acid sequence we identified a linker of ∼20 residues between the second transmembrane helix and the cytosolic domain. This unique polypeptide appears to be highly flexible and turned out to be essential for proper functioning of FtsH as its deletion fully eliminated the proteolytic activity of FtsH.
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 マップデータ
マップデータ 試料
試料
 データ登録者
データ登録者 オランダ,
オランダ,  スイス, 2件
スイス, 2件  引用
引用 構造の表示
構造の表示 ムービービューア
ムービービューア
 ダウンロードとリンク
ダウンロードとリンク emd_11167.png
emd_11167.png http://ftp.pdbj.org/pub/emdb/structures/EMD-11167
http://ftp.pdbj.org/pub/emdb/structures/EMD-11167












 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)


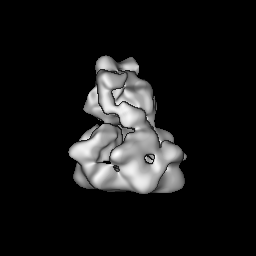

















 試料の構成要素
試料の構成要素 解析
解析 電子顕微鏡法
電子顕微鏡法 FIELD EMISSION GUN
FIELD EMISSION GUN