 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-11011 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Localized reconstruction of human adenovirus D26 hexon type 2 | |||||||||
 マップデータ マップデータ | Localized reconstruction of human adenovirus D26 hexon 2 | |||||||||
 試料 試料 |
| |||||||||
| 生物種 |  Homo sapiens (ヒト) Homo sapiens (ヒト) | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 3.1 Å | |||||||||
 データ登録者 データ登録者 | Huiskonen JT / Abrishami V | |||||||||
| 資金援助 | European Union,  英国, 2件 英国, 2件
| |||||||||
 引用 引用 | ジャーナル: Prog Biophys Mol Biol / 年: 2021 タイトル: Localized reconstruction in Scipion expedites the analysis of symmetry mismatches in cryo-EM data. 著者: Vahid Abrishami / Serban L Ilca / Josue Gomez-Blanco / Ilona Rissanen / José Miguel de la Rosa-Trevín / Vijay S Reddy / José-Maria Carazo / Juha T Huiskonen /      要旨: Technological advances in transmission electron microscopes and detectors have turned cryogenic electron microscopy (cryo-EM) into an essential tool for structural biology. A commonly used cryo-EM ...Technological advances in transmission electron microscopes and detectors have turned cryogenic electron microscopy (cryo-EM) into an essential tool for structural biology. A commonly used cryo-EM data analysis method, single particle analysis, averages hundreds of thousands of low-dose images of individual macromolecular complexes to determine a density map of the complex. The presence of symmetry in the complex is beneficial since each projection image can be assigned to multiple views of the complex. However, data processing that applies symmetry can average out asymmetric features and consequently data analysis methods are required to resolve asymmetric structural features. Scipion is a cryo-EM image processing framework that integrates functions from different image processing packages as plugins. To extend its functionality for handling symmetry mismatches, we present here a Scipion plugin termed LocalRec implementing the localized reconstruction method. When tested on an adenovirus data set, the plugin enables resolving the symmetry-mismatched trimeric fibre bound to the five-fold vertices of the capsid. Furthermore, it improves the structure determination of the icosahedral capsid by dealing with the defocus gradient across the particle. LocalRec is expected to be widely applicable in a range of cryo-EM investigations of flexible and symmetry mismatched complexes. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
 ムービービューア ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |
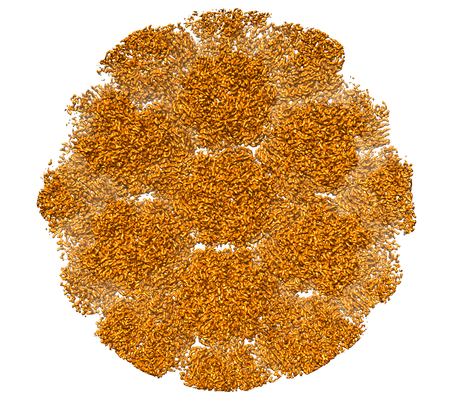
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_11011.map.gz | 27 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-11011-v30.xmlemd-11011.xml | 17.2 KB 17.2 KB | 表示 表示 | EMDBヘッダ |
| FSC (解像度算出) | emd_11011_fsc.xml | 9.1 KB | 表示 | FSCデータファイル |
| 画像 | 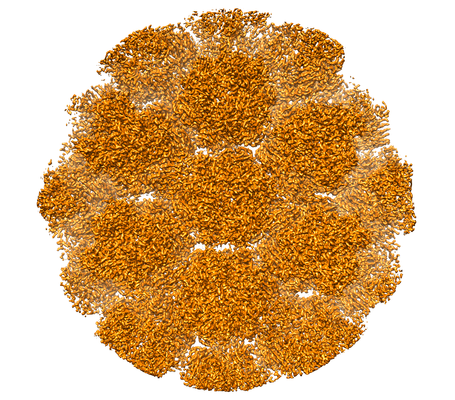 emd_11011.png emd_11011.png | 299.8 KB | ||
| マスクデータ | emd_11011_msk_1.map | 64 MB | マスクマップ | |
| その他 | emd_11011_half_map_1.map.gzemd_11011_half_map_2.map.gz | 48.8 MB 48.8 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-11011ftp://ftp.pdbj.org/pub/emdb/structures/EMD-11011 http://ftp.pdbj.org/pub/emdb/structures/EMD-11011ftp://ftp.pdbj.org/pub/emdb/structures/EMD-11011 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_11011_validation.pdf.gz | 542.9 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_11011_full_validation.pdf.gz | 542 KB | 表示 | |
| XML形式データ | emd_11011_validation.xml.gz | 14.8 KB | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-11011ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-11011 | HTTPS FTP |
-関連構造データ
| 関連構造データ | C: 同じ文献を引用 ( |
|---|---|
| 類似構造データ | |
| 電子顕微鏡画像生データ | EMPIAR-10455 (タイトル: Single particle cryo-EM dataset of human adenovirus HAdV-D26 Data size: 46.5 Data #1: Particle stacks of human adenovirus HAdV-D26 [picked particles - single frame - processed]) |








-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_11011.map.gz / 形式: CCP4 / 大きさ: 64 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Localized reconstruction of human adenovirus D26 hexon 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.31 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-添付データ
-マスク #1
| ファイル | emd_11011_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |




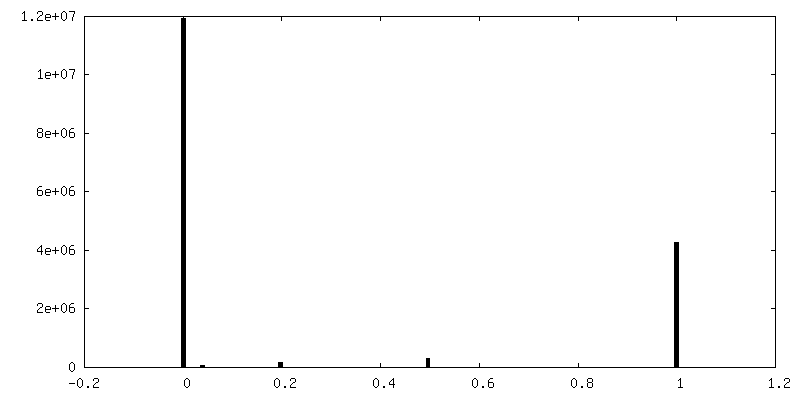
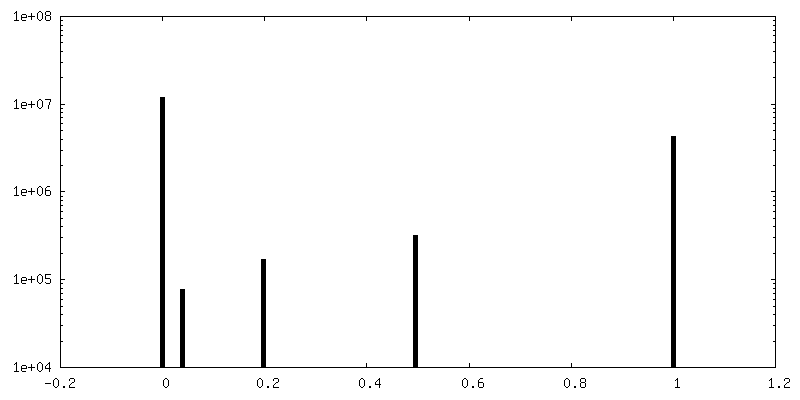
-ハーフマップ: Half 2 of human adenovirus D26 hexon 2 map
| ファイル | emd_11011_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Half 2 of human adenovirus D26 hexon 2 map | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |







-ハーフマップ: Half 1 of human adenovirus D26 hexon 2 map
| ファイル | emd_11011_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Half 1 of human adenovirus D26 hexon 2 map | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |




- 試料の構成要素
試料の構成要素
-全体 : Adenovirus hexon type 2
| 全体 | 名称: Adenovirus hexon type 2 |
|---|---|
| 要素 |
|
-超分子 #1: Adenovirus hexon type 2
| 超分子 | 名称: Adenovirus hexon type 2 / タイプ: complex / ID: 1 / 親要素: 0 |
|---|---|
| 由来(天然) | 生物種: Homo sapiens (ヒト) |
| 組換発現 | 生物種: Homo sapiens (ヒト) / 組換細胞: HEK293 |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 6 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 緩衝液 | pH: 8.1 構成要素:
| ||||||||||||
| 凍結 | 凍結剤: ETHANE / 装置: GATAN CRYOPLUNGE 3 |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 撮影 | フィルム・検出器のモデル: GATAN K2 SUMMIT (4k x 4k) 検出モード: COUNTING / デジタル化 - 画像ごとのフレーム数: 7-38 / 実像数: 2000 / 平均露光時間: 7.6 sec. / 平均電子線量: 53.0 e/Å2 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 照射モード: OTHER / 撮影モード: OTHER / Cs: 2.7 mm / 最大 デフォーカス(公称値): 3.0 µm / 最小 デフォーカス(公称値): 0.8 µm / 倍率(公称値): 22500 |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |


