Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-10206 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of the curli secretion-assembly complex CsgG:CsgF | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Secretion Channel / Curli / Outer Membrane Protein / Nanopore Sensing / Protein Transport / Bacterial amyloid | |||||||||
| Function / homology |  Function and homology information Function and homology informationcurli secretion complex / curli assembly / protein secretion by the type VIII secretion system / protein transmembrane transport / single-species biofilm formation / cell outer membrane / outer membrane-bounded periplasmic space / identical protein binding / plasma membrane Similarity search - Function | |||||||||
| Biological species |  | |||||||||
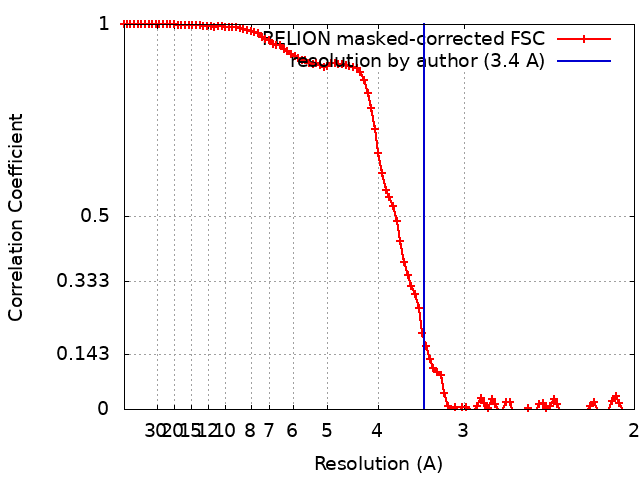
| Method | single particle reconstruction / cryo EM / Resolution: 3.4 Å | |||||||||
 Authors Authors | Van der Verren SE / Remaut H | |||||||||
| Funding support |  Belgium, 2 items Belgium, 2 items
| |||||||||
 Citation Citation | Journal: Nat Biotechnol / Year: 2020 Title: A dual-constriction biological nanopore resolves homonucleotide sequences with high fidelity. Authors: Sander E Van der Verren / Nani Van Gerven / Wim Jonckheere / Richard Hambley / Pratik Singh / John Kilgour / Michael Jordan / E Jayne Wallace / Lakmal Jayasinghe / Han Remaut /  Abstract: Single-molecule long-read DNA sequencing with biological nanopores is fast and high-throughput but suffers reduced accuracy in homonucleotide stretches. We now combine the CsgG nanopore with the 35- ...Single-molecule long-read DNA sequencing with biological nanopores is fast and high-throughput but suffers reduced accuracy in homonucleotide stretches. We now combine the CsgG nanopore with the 35-residue N-terminal region of its extracellular interaction partner CsgF to produce a dual-constriction pore with improved signal and base-calling accuracy for homopolymer regions. The electron cryo-microscopy structure of CsgG in complex with full-length CsgF shows that the 33 N-terminal residues of CsgF bind inside the β-barrel of the pore, forming a defined second constriction. In complexes of CsgG bound to a 35-residue CsgF constriction peptide, the second constriction is separated from the primary constriction by ~25 Å. We find that both constrictions contribute to electrical signal modulation during single-stranded DNA translocation. DNA sequencing using a prototype CsgG-CsgF protein pore with two constrictions improved single-read accuracy by 25 to 70% in homopolymers up to 9 nucleotides long. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_10206.map.gz | 10.6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-10206-v30.xmlemd-10206.xml | 14.2 KB 14.2 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_10206_fsc.xml | 9.9 KB | Display | FSC data file |
| Images |  emd_10206.png emd_10206.png | 126.2 KB | ||
| Filedesc metadata | emd-10206.cif.gz | 6 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-10206ftp://ftp.pdbj.org/pub/emdb/structures/EMD-10206 http://ftp.pdbj.org/pub/emdb/structures/EMD-10206ftp://ftp.pdbj.org/pub/emdb/structures/EMD-10206 | HTTPS FTP |
-Related structure data
| Related structure data |  6si7MC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |





-Map
| File | Download / File: emd_10206.map.gz / Format: CCP4 / Size: 83.7 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : CsgG:CsgF complex in DDM
| Entire | Name: CsgG:CsgF complex in DDM |
|---|---|
| Components |
|
-Supramolecule #1: CsgG:CsgF complex in DDM
| Supramolecule | Name: CsgG:CsgF complex in DDM / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 400 KDa |
-Macromolecule #1: Curli production assembly/transport component CsgF
| Macromolecule | Name: Curli production assembly/transport component CsgF / type: protein_or_peptide / ID: 1 / Details: Only first 35 residues were visible and built / Number of copies: 9 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 13.744857 KDa |
| Recombinant expression | Organism: |
| Sequence | String: GTMTFQFRNP NFGGNPNNGA FLLNSAQAQN SYKDPSYNDD FGIETPSALD NFTQAIQSQI LGGLLSNINT GKPGRMVTND YIVDIANRD GQLQLNVTDR KTGQTSTIQV SGLQNNSTDF HHHHHH UniProtKB: Curli production assembly/transport component CsgF |
-Macromolecule #2: Curli production assembly/transport component CsgG
| Macromolecule | Name: Curli production assembly/transport component CsgG / type: protein_or_peptide / ID: 2 / Number of copies: 9 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 30.110193 KDa |
| Recombinant expression | Organism: |
| Sequence | String: CLTAPPKEAA RPTLMPRAQS YKDLTHLPAP TGKIFVSVYN IQDETGQFKP YPASNFSTAV PQSATAMLVT ALKDSRWFIP LERQGLQNL LNERKIIRAA QENGTVAINN RIPLQSLTAA NIMVEGSIIG YESNVKSGGV GARYFGIGAD TQYQLDQIAV N LRVVNVST ...String: CLTAPPKEAA RPTLMPRAQS YKDLTHLPAP TGKIFVSVYN IQDETGQFKP YPASNFSTAV PQSATAMLVT ALKDSRWFIP LERQGLQNL LNERKIIRAA QENGTVAINN RIPLQSLTAA NIMVEGSIIG YESNVKSGGV GARYFGIGAD TQYQLDQIAV N LRVVNVST GEILSSVNTS KTILSYEVQA GVFRFIDYQR LLEGEVGYTS NEPVMLCLMS AIETGVIFLI NDGIDRGLWD LQ NKAERQN DILVKYRHMS VPPESSAWSH PQFEK UniProtKB: Curli production assembly/transport component CsgG |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.03 mg/mL | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 8 Component:
| ||||||||
| Grid | Model: Quantifoil R2/1 / Material: COPPER / Mesh: 400 / Support film - Material: GRAPHENE OXIDE / Support film - topology: HOLEY | ||||||||
| Vitrification | Cryogen name: ETHANE / Instrument: GATAN CRYOPLUNGE 3 |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Number real images: 2045 / Average electron dose: 56.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal magnification: 130000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |