 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-10162: Cryo-EM structure of the ClpXP1/2 protein degradation machinery -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-10162 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of the ClpXP1/2 protein degradation machinery | ||||||||||||
 Map data Map data | CryoEM density of ClpXP1/2 from Listeria monocytogenes. ClpP1/2 density filtered to 3.9A. ClpX density filtered to 6A. | ||||||||||||
 Sample Sample |
| ||||||||||||
| Function / homology |  Function and homology information Function and homology informationHslUV protease complex / endopeptidase Clp / endopeptidase Clp complex / ATP-dependent peptidase activity / protein quality control for misfolded or incompletely synthesized proteins / proteolysis involved in protein catabolic process / ATP-dependent protein folding chaperone / unfolded protein binding / peptidase activity / ATPase binding ...HslUV protease complex / endopeptidase Clp / endopeptidase Clp complex / ATP-dependent peptidase activity / protein quality control for misfolded or incompletely synthesized proteins / proteolysis involved in protein catabolic process / ATP-dependent protein folding chaperone / unfolded protein binding / peptidase activity / ATPase binding / protein dimerization activity / serine-type endopeptidase activity / cell division / ATP hydrolysis activity / proteolysis / zinc ion binding / ATP binding / cytoplasm Similarity search - Function | ||||||||||||
| Biological species |  Listeria monocytogenes s (bacteria) / Listeria monocytogenes EGD (bacteria) Listeria monocytogenes s (bacteria) / Listeria monocytogenes EGD (bacteria) | ||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 4.0 Å | ||||||||||||
 Authors Authors | Gatsogiannis C / Merino F / Raunser S | ||||||||||||
| Funding support |  Germany, 3 items Germany, 3 items
| ||||||||||||
 Citation Citation | Journal: Nat Struct Mol Biol / Year: 2019 Title: Cryo-EM structure of the ClpXP protein degradation machinery. Authors: Christos Gatsogiannis / Dora Balogh / Felipe Merino / Stephan A Sieber / Stefan Raunser / Abstract: The ClpXP machinery is a two-component protease complex that performs targeted protein degradation in bacteria and mitochondria. The complex consists of the AAA+ chaperone ClpX and the peptidase ClpP. ...The ClpXP machinery is a two-component protease complex that performs targeted protein degradation in bacteria and mitochondria. The complex consists of the AAA+ chaperone ClpX and the peptidase ClpP. The hexameric ClpX utilizes the energy of ATP binding and hydrolysis to engage, unfold and translocate substrates into the catalytic chamber of tetradecameric ClpP, where they are degraded. Formation of the complex involves a symmetry mismatch, because hexameric AAA+ rings bind axially to the opposing stacked heptameric rings of the tetradecameric ClpP. Here we present the cryo-EM structure of ClpXP from Listeria monocytogenes. We unravel the heptamer-hexamer binding interface and provide novel insight into the ClpX-ClpP cross-talk and activation mechanism. Comparison with available crystal structures of ClpP and ClpX in different states allows us to understand important aspects of the complex mode of action of ClpXP and provides a structural framework for future pharmacological applications. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_10162.map.gz | 39.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-10162-v30.xmlemd-10162.xml | 22.1 KB 22.1 KB | Display Display | EMDB header |
| Images |  emd_10162.png emd_10162.png | 109.2 KB | ||
| Others | emd_10162_half_map_1.map.gzemd_10162_half_map_2.map.gz | 23 MB 23 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-10162ftp://ftp.pdbj.org/pub/emdb/structures/EMD-10162 http://ftp.pdbj.org/pub/emdb/structures/EMD-10162ftp://ftp.pdbj.org/pub/emdb/structures/EMD-10162 | HTTPS FTP |
-Validation report
| Summary document | emd_10162_validation.pdf.gz | 386.5 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_10162_full_validation.pdf.gz | 385.7 KB | Display | |
| Data in XML | emd_10162_validation.xml.gz | 10.2 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-10162ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-10162 | HTTPS FTP |
-Related structure data
| Related structure data |  6sfwMC  6sfxMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_10162.map.gz / Format: CCP4 / Size: 42.9 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | CryoEM density of ClpXP1/2 from Listeria monocytogenes. ClpP1/2 density filtered to 3.9A. ClpX density filtered to 6A. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
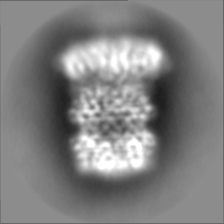
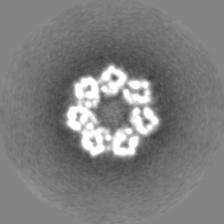
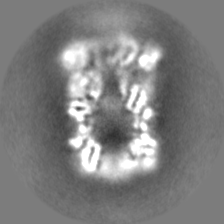
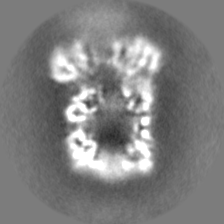
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.09 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Half map: half map 1
| File | emd_10162_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
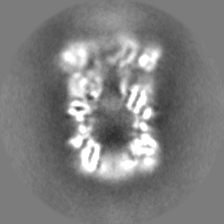
| Density Histograms |
-Half map: half map 0
| File | emd_10162_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map 0 | ||||||||||||
| Projections & Slices |
| ||||||||||||
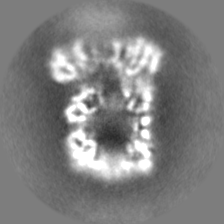
| Density Histograms |


- Sample components
Sample components
-Entire : LmClpXP1/2 heterocomplex
| Entire | Name: LmClpXP1/2 heterocomplex |
|---|---|
| Components |
|
-Supramolecule #1: LmClpXP1/2 heterocomplex
| Supramolecule | Name: LmClpXP1/2 heterocomplex / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Listeria monocytogenes s (bacteria) |
| Recombinant expression | Organism: |
-Supramolecule #2: LmClpXP1/2
| Supramolecule | Name: LmClpXP1/2 / type: complex / ID: 2 / Parent: 1 / Macromolecule list: all Details: LmCLP1 and LmCLP2 assemble into two heptameric rings which stack face to face to form the LmClpP1/2 heterocomplex. The chaperone ClpX forms a hexamer, that associates with the LmClpP2 heptamer. |
|---|---|
| Source (natural) | Organism: Listeria monocytogenes EGD (bacteria) |
| Recombinant expression | Organism: |
-Macromolecule #1: LmClpP1
| Macromolecule | Name: LmClpP1 / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO / EC number: endopeptidase Clp |
|---|---|
| Source (natural) | Organism: Listeria monocytogenes EGD (bacteria) |
| Recombinant expression | Organism: |
| Sequence | String: MAENTKNENI TNILTQKLID TRTVLIYGEI NQELAEDVSK QLLLLESISN DPITIFINSQ GGHVEAGDTI HDMIKFIKPT VKVVGTGWV ASAGITIYLA AEKENRFSLP NTRYMIHQPA GGVQGQSTEI EIEAKEIIRM RERINRLIAE ATGQSYEQIS K DTDRNFWL ...String: MAENTKNENI TNILTQKLID TRTVLIYGEI NQELAEDVSK QLLLLESISN DPITIFINSQ GGHVEAGDTI HDMIKFIKPT VKVVGTGWV ASAGITIYLA AEKENRFSLP NTRYMIHQPA GGVQGQSTEI EIEAKEIIRM RERINRLIAE ATGQSYEQIS K DTDRNFWL SVNEAKDYGI VNEIIENRDG LKMASWSHPQ FEK |
-Macromolecule #2: LmClpP2
| Macromolecule | Name: LmClpP2 / type: protein_or_peptide / ID: 2 / Enantiomer: LEVO / EC number: endopeptidase Clp |
|---|---|
| Source (natural) | Organism: Listeria monocytogenes EGD (bacteria) |
| Recombinant expression | Organism: |
| Sequence | String: MNLIPTVIEQ TSRGERAYDI YSRLLKDRII MLGSAIDDNV ANSIVSQLLF LDAQDPEKDI FLYINSPGGS ISAGMAIYDT MNFVKADVQ TIGMGMAASM GSFLLTAGAN GKRFALPNAE IMIHQPLGGA QGQATEIEIA ARHILKIKER MNTIMAEKTG Q PYEVIARD ...String: MNLIPTVIEQ TSRGERAYDI YSRLLKDRII MLGSAIDDNV ANSIVSQLLF LDAQDPEKDI FLYINSPGGS ISAGMAIYDT MNFVKADVQ TIGMGMAASM GSFLLTAGAN GKRFALPNAE IMIHQPLGGA QGQATEIEIA ARHILKIKER MNTIMAEKTG Q PYEVIARD TDRDNFMTAQ EAKDYGLIDD IIINKSGLKG HHHHHH DTD RNFWLSVNEA KDYGIVNEII ENRDGLKMAS WSHPQFEK |
-Macromolecule #3: LmClpX
| Macromolecule | Name: LmClpX / type: protein_or_peptide / ID: 3 / Enantiomer: LEVO / EC number: endopeptidase Clp |
|---|---|
| Source (natural) | Organism: Listeria monocytogenes EGD (bacteria) |
| Recombinant expression | Organism: |
| Sequence | String: MFKFNDEKGQ LKCSFCGKTQ DQVRKLVAGP GVYICDECIE LCNEIIEEEL GISEFVDFGE VPKPQEIRHI LSDYVIGQE RAKKALAVAV YNHYKRINSN ETKEDEVELS KSNICLIGPT GSGKTLLAQT LARILNVPFA I ADATSLTE AGYVGEDVEN ILLKLIQSAD ...String: MFKFNDEKGQ LKCSFCGKTQ DQVRKLVAGP GVYICDECIE LCNEIIEEEL GISEFVDFGE VPKPQEIRHI LSDYVIGQE RAKKALAVAV YNHYKRINSN ETKEDEVELS KSNICLIGPT GSGKTLLAQT LARILNVPFA I ADATSLTE AGYVGEDVEN ILLKLIQSAD YDVEKAEKGI IYIDEIDKVA RKSENPSITR DVSGEGVQQA LL KILEGTV ASVPPQGGRK HPHQELIQID TGNILFIVGG AFDGIEQIVK NRMGEKVIGF GTDNAKLKDD ETY LSRVVP EDLLKFGLIP EFIGRLPVIA TLEQLDEAAL VSILTEPKNA LVKQYKRMLE LDDVELEFEP TALI EIAKE AIERKTGARG LRSIIEQIML EVMFEIPSRD DITKCIITEK AARGEEEPQL QLEDGSIIPI KTSA |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.01 mg/mL | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.6 Component:
Details: The sample was crosslinked with 0.1% glutaraldehyde. The reaction was quenched after 30sec with 2 eq. Tris-HCL. | ||||||||||||||
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 400 / Support film - Material: CARBON / Support film - topology: CONTINUOUS / Support film - Film thickness: 2.0 nm / Pretreatment - Type: GLOW DISCHARGE | ||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 90 % / Instrument: GATAN CRYOPLUNGE 3 Details: the sample was blotted after 45sec of incubation time.. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Detector mode: INTEGRATING / Number grids imaged: 1 / Number real images: 3200 / Average exposure time: 2.0 sec. / Average electron dose: 114.0 e/Å2 Details: Images were collected in movie-mode at a frame rate of 50msec |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 112807 / Illumination mode: OTHER / Imaging mode: BRIGHT FIELD / Cs: 0.0 mm / Nominal magnification: 59000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
