Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-0409: Single-particle cryo-EM reconstruction of the catalytic subunit o... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-0409 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Single-particle cryo-EM reconstruction of the catalytic subunit of protein kinase A bound to ATP and IP20 using 200 keV | |||||||||
 Map data Map data | Single-particle cryo-EM reconstruction of the catalytic subunit of protein kinase A bound to ATP and IP20 (unsharpened) | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  | |||||||||
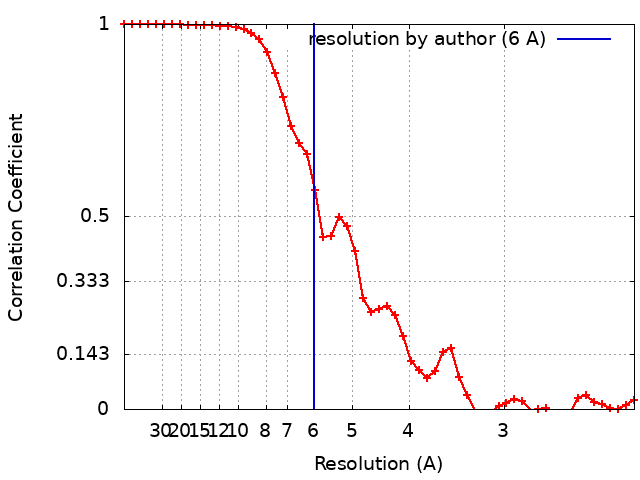
| Method | single particle reconstruction / cryo EM / Resolution: 6.0 Å | |||||||||
 Authors Authors | Herzik Jr MA / Wu M / Lander GC | |||||||||
 Citation Citation | Journal: Nat Commun / Year: 2019 Title: High-resolution structure determination of sub-100 kDa complexes using conventional cryo-EM. Authors: Mark A Herzik / Mengyu Wu / Gabriel C Lander /  Abstract: Determining high-resolution structures of biological macromolecules amassing less than 100 kilodaltons (kDa) has been a longstanding goal of the cryo-electron microscopy (cryo-EM) community. While ...Determining high-resolution structures of biological macromolecules amassing less than 100 kilodaltons (kDa) has been a longstanding goal of the cryo-electron microscopy (cryo-EM) community. While the Volta phase plate has enabled visualization of specimens in this size range, this instrumentation is not yet fully automated and can present technical challenges. Here, we show that conventional defocus-based cryo-EM methodologies can be used to determine high-resolution structures of specimens amassing less than 100 kDa using a transmission electron microscope operating at 200 keV coupled with a direct electron detector. Our ~2.7 Å structure of alcohol dehydrogenase (82 kDa) proves that bound ligands can be resolved with high fidelity to enable investigation of drug-target interactions. Our ~2.8 Å and ~3.2 Å structures of methemoglobin demonstrate that distinct conformational states can be identified within a dataset for proteins as small as 64 kDa. Furthermore, we provide the sub-nanometer cryo-EM structure of a sub-50 kDa protein. | |||||||||
| History |
|
- Structure visualization
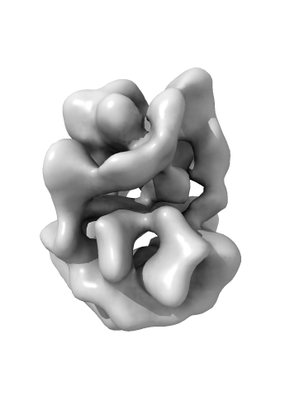
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_0409.map.gz | 6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-0409-v30.xmlemd-0409.xml | 21.2 KB 21.2 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_0409_fsc.xml | 5.4 KB | Display | FSC data file |
| Images |  emd_0409.png emd_0409.png | 35.8 KB | ||
| Others | emd_0409_additional.map.gzemd_0409_half_map_1.map.gzemd_0409_half_map_2.map.gz | 7.4 MB 6 MB 6 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-0409ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0409 http://ftp.pdbj.org/pub/emdb/structures/EMD-0409ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0409 | HTTPS FTP |
-Related structure data
| Related structure data |  0406C  0407C  0408C  6nbbC  6nbcC  6nbdC C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10252 (Title: Catalytic subunit of protein kinase A bound to ATP, manganese, and IP20 movies obtained using Talos Arctica operating at 200 kV equipped with a K2 Data size: 5.5 TB Data #1: Raw, unaligned movie stacks of the catalytic subunit of protein kinase A bound to ATP, manganese, and IP20 acquired on a Talos Arctica using a K2 direct electron detector [micrographs - multiframe]) |








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_0409.map.gz / Format: CCP4 / Size: 8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Single-particle cryo-EM reconstruction of the catalytic subunit of protein kinase A bound to ATP and IP20 (unsharpened) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.1172 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
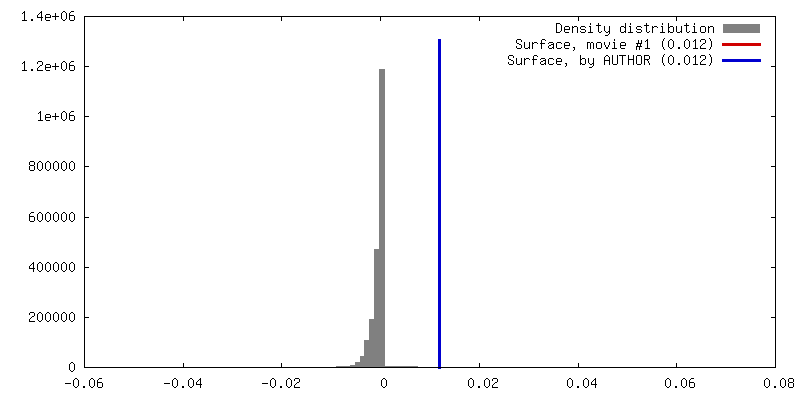
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
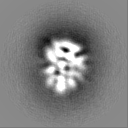
-Additional map: Single-particle cryo-EM reconstruction of the catalytic subunit of...
| File | emd_0409_additional.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Single-particle cryo-EM reconstruction of the catalytic subunit of protein kinase A bound to ATP and IP20 (sharpened) | ||||||||||||
| Projections & Slices |
| ||||||||||||
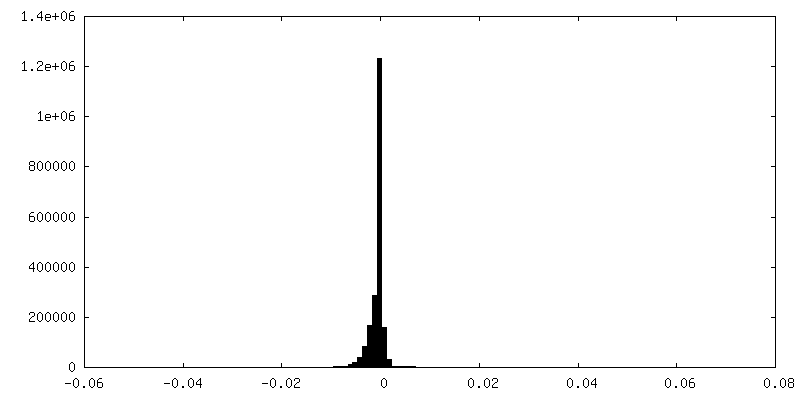
| Density Histograms |






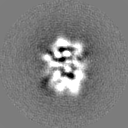
-Half map: Odd half map
| File | emd_0409_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Odd half map | ||||||||||||
| Projections & Slices |
| ||||||||||||
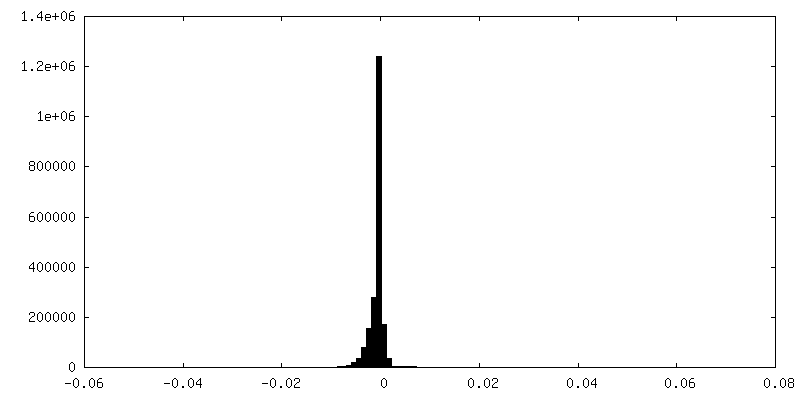
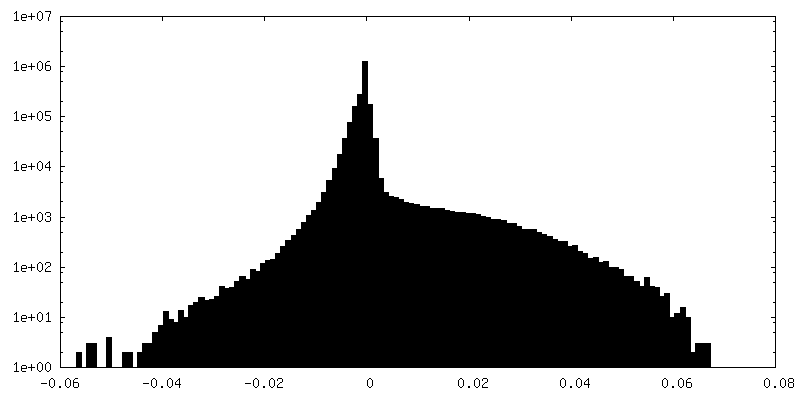
| Density Histograms |



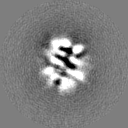
-Half map: Even half map
| File | emd_0409_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Even half map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |


- Sample components
Sample components
-Entire : catalytic subunit of mouse protein kinase A bound to ATP and IP20
| Entire | Name: catalytic subunit of mouse protein kinase A bound to ATP and IP20 |
|---|---|
| Components |
|
-Supramolecule #1: catalytic subunit of mouse protein kinase A bound to ATP and IP20
| Supramolecule | Name: catalytic subunit of mouse protein kinase A bound to ATP and IP20 type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism:  |
| Molecular weight | Theoretical: 43 KDa |
-Macromolecule #1: catalytic subunit of protein kinase A
| Macromolecule | Name: catalytic subunit of protein kinase A / type: protein_or_peptide / ID: 1 / Enantiomer: LEVO / EC number: non-specific serine/threonine protein kinase |
|---|---|
| Source (natural) | Organism: |
| Recombinant expression | Organism: |
| Sequence | String: GNAAAAKKGS EQESVKEFLA KAKEDFLKKW ETPSQNTAQL DQFDRIKTLG TGSFGRVMLV KHKESGNHYA MKILDKQKVV KLKQIEHTL NEKRILQAVN FPFLVKLEFS FKDNSNLYMV MEYVAGGEMF SHLRRIGRFS EPHARFYAAQ IVLTFEYLHS L DLIYRDLK ...String: GNAAAAKKGS EQESVKEFLA KAKEDFLKKW ETPSQNTAQL DQFDRIKTLG TGSFGRVMLV KHKESGNHYA MKILDKQKVV KLKQIEHTL NEKRILQAVN FPFLVKLEFS FKDNSNLYMV MEYVAGGEMF SHLRRIGRFS EPHARFYAAQ IVLTFEYLHS L DLIYRDLK PENLLIDQQG YIQVTDFGFA KRVKGRTWTL CGTPEYLAPE IILSKGYNKA VDWWALGVLI YEMAAGYPPF FA DQPIQIY EKIVSGKVRF PSHFSSDLKD LLRNLLQVDL TKRFGNLKNG VNDIKNHKWF ATTDWIAIYQ RKVEAPFIPK FKG PGDTSN FDDYEEEEIR VSINEKCGKE FTEF |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 5 mg/mL | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7 Component:
| |||||||||||||||||||||
| Grid | Model: Quantifoil, UltrAuFoil, R1.2/1.3 / Material: GOLD / Mesh: 300 / Pretreatment - Type: PLASMA CLEANING / Pretreatment - Atmosphere: OTHER Details: Grids were plasma cleaned using a Solarus plasma cleaner (Gatan, Inc.) | |||||||||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 90 % / Chamber temperature: 277.15 K / Instrument: HOMEMADE PLUNGER Details: Sample was manually blotted for 4-5 seconds using Whatman No. 1 filter paper immediately prior to plunge-freezing.. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TALOS ARCTICA |
|---|---|
| Image recording | #0 - Image recording ID: 1 / #0 - Film or detector model: GATAN K2 SUMMIT (4k x 4k) / #0 - Detector mode: COUNTING / #0 - Number grids imaged: 2 / #0 - Number real images: 3176 / #0 - Average exposure time: 11.0 sec. / #0 - Average electron dose: 69.0 e/Å2 / #1 - Image recording ID: 2 / #1 - Film or detector model: GATAN K2 SUMMIT (4k x 4k) / #1 - Detector mode: COUNTING / #1 - Digitization - Dimensions - Width: 3710 pixel / #1 - Digitization - Dimensions - Height: 3838 pixel / #1 - Digitization - Sampling interval: 5.0 µm / #1 - Digitization - Frames/image: 1-44 / #1 - Number grids imaged: 2 / #1 - Number real images: 2402 / #1 - Average exposure time: 11.0 sec. / #1 - Average electron dose: 69.0 e/Å2 / #1 - Details: Specimen was tilted at 30 degrees |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 16.0 µm / Nominal defocus min: 5.0 µm / Nominal magnification: 73000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Talos Arctica / Image courtesy: FEI Company |