 Movie
Movie Controller
Controller
[English] 日本語
 Yorodumi
Yorodumi- SASDEZ2: Unlabeled nuclear pore complex protein Nup153 (NUL) without denaturant -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: SASBDB / ID: SASDEZ2 |
|---|---|
 Sample Sample | Unlabeled nuclear pore complex protein Nup153 (NUL) without denaturant
|
| Function / homology |  Function and homology information Function and homology informationnegative regulation of RNA export from nucleus / nuclear pore complex assembly / Nuclear Pore Complex (NPC) Disassembly / Regulation of Glucokinase by Glucokinase Regulatory Protein / Defective TPR may confer susceptibility towards thyroid papillary carcinoma (TPC) / nuclear inclusion body / Transport of Ribonucleoproteins into the Host Nucleus / nuclear pore nuclear basket / Transport of the SLBP independent Mature mRNA / Transport of the SLBP Dependant Mature mRNA ...negative regulation of RNA export from nucleus / nuclear pore complex assembly / Nuclear Pore Complex (NPC) Disassembly / Regulation of Glucokinase by Glucokinase Regulatory Protein / Defective TPR may confer susceptibility towards thyroid papillary carcinoma (TPC) / nuclear inclusion body / Transport of Ribonucleoproteins into the Host Nucleus / nuclear pore nuclear basket / Transport of the SLBP independent Mature mRNA / Transport of the SLBP Dependant Mature mRNA / NS1 Mediated Effects on Host Pathways / SUMOylation of SUMOylation proteins / structural constituent of nuclear pore / Transport of Mature mRNA Derived from an Intronless Transcript / Rev-mediated nuclear export of HIV RNA / Nuclear import of Rev protein / SUMOylation of RNA binding proteins / NEP/NS2 Interacts with the Cellular Export Machinery / RNA export from nucleus / Transport of Mature mRNA derived from an Intron-Containing Transcript / tRNA processing in the nucleus / nuclear localization sequence binding / Viral Messenger RNA Synthesis / nucleocytoplasmic transport / SUMOylation of ubiquitinylation proteins / Vpr-mediated nuclear import of PICs / SUMOylation of DNA replication proteins / Regulation of HSF1-mediated heat shock response / nuclear pore / mRNA transport / SUMOylation of DNA damage response and repair proteins / protein-membrane adaptor activity / nuclear periphery / SUMOylation of chromatin organization proteins / HCMV Late Events / molecular condensate scaffold activity / Transcriptional regulation by small RNAs / viral penetration into host nucleus / ISG15 antiviral mechanism / HCMV Early Events / protein import into nucleus / nuclear envelope / host cell / snRNP Assembly / nuclear membrane / amyloid fibril formation / symbiont entry into host cell / nucleolus / SARS-CoV-2 activates/modulates innate and adaptive immune responses / DNA binding / zinc ion binding / nucleoplasm / membrane / identical protein binding / cytosol Similarity search - Function |
| Biological species |  Homo sapiens (human) Homo sapiens (human) |
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2017 Title: Decoupling of size and shape fluctuations in heteropolymeric sequences reconciles discrepancies in SAXS vs. FRET measurements. Authors: Gustavo Fuertes / Niccolò Banterle / Kiersten M Ruff / Aritra Chowdhury / Davide Mercadante / Christine Koehler / Michael Kachala / Gemma Estrada Girona / Sigrid Milles / Ankur Mishra / ...Authors: Gustavo Fuertes / Niccolò Banterle / Kiersten M Ruff / Aritra Chowdhury / Davide Mercadante / Christine Koehler / Michael Kachala / Gemma Estrada Girona / Sigrid Milles / Ankur Mishra / Patrick R Onck / Frauke Gräter / Santiago Esteban-Martín / Rohit V Pappu / Dmitri I Svergun / Edward A Lemke /     Abstract: Unfolded states of proteins and native states of intrinsically disordered proteins (IDPs) populate heterogeneous conformational ensembles in solution. The average sizes of these heterogeneous ...Unfolded states of proteins and native states of intrinsically disordered proteins (IDPs) populate heterogeneous conformational ensembles in solution. The average sizes of these heterogeneous systems, quantified by the radius of gyration ( ), can be measured by small-angle X-ray scattering (SAXS). Another parameter, the mean dye-to-dye distance ( ) for proteins with fluorescently labeled termini, can be estimated using single-molecule Förster resonance energy transfer (smFRET). A number of studies have reported inconsistencies in inferences drawn from the two sets of measurements for the dimensions of unfolded proteins and IDPs in the absence of chemical denaturants. These differences are typically attributed to the influence of fluorescent labels used in smFRET and to the impact of high concentrations and averaging features of SAXS. By measuring the dimensions of a collection of labeled and unlabeled polypeptides using smFRET and SAXS, we directly assessed the contributions of dyes to the experimental values and For chemically denatured proteins we obtain mutual consistency in our inferences based on and , whereas for IDPs under native conditions, we find substantial deviations. Using computations, we show that discrepant inferences are neither due to methodological shortcomings of specific measurements nor due to artifacts of dyes. Instead, our analysis suggests that chemical heterogeneity in heteropolymeric systems leads to a decoupling between and that is amplified in the absence of denaturants. Therefore, joint assessments of and combined with measurements of polymer shapes should provide a consistent and complete picture of the underlying ensembles. |
 Contact author Contact author |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Data source
| SASBDB page |  SASDEZ2 SASDEZ2 |
|---|
-Related structure data
| Related structure data | C: citing same article ( |
|---|---|
| Similar structure data |










-External links
| Related items in Molecule of the Month |
|---|



















-Models
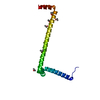
| Model #2291 |   Type: atomic Comment: p-acetylphenylalanine was replaced with a cysteine. Chi-square value: 1.927 / P-value: 0.000012  Search similar-shape structures of this assembly by Omokage search (details) Search similar-shape structures of this assembly by Omokage search (details) |
|---|---|
| Model #2292 |  Type: atomic Comment: p-acetylphenylalanine was replaced with a cysteine. Chi-square value: 1.927 / P-value: 0.000012 Search similar-shape structures of this assembly by Omokage search (details) |
| Model #2293 |  Type: atomic / Software: (CAMPARI) Comment: p-acetylphenylalanine was replaced with a cysteine. Chi-square value: 1.927 / P-value: 0.000012 Search similar-shape structures of this assembly by Omokage search (details) |
| Model #2294 |  Type: atomic Comment: p-acetylphenylalanine was replaced with a cysteine. Chi-square value: 1.927 / P-value: 0.000012 Search similar-shape structures of this assembly by Omokage search (details) |
-Sample
| Sample | Name: Unlabeled nuclear pore complex protein Nup153 (NUL) without denaturant Specimen concentration: 2.00-5.00 |
|---|---|
| Buffer | Name: PBS, 10 mM DTT / pH: 7.4 |
| Entity #1240 | Name: NUL / Type: protein / Description: Nuclear pore complex protein Nup153 / Formula weight: 11.717 / Num. of mol.: 1 / Source: Homo sapiens / References: UniProt: P49790 Sequence: GCGFKGFDTS SSSSNSAASS SFKFGVSSSS SGPSQTLTST GNFKFGDQGG FKIGVSSDSG SINPMSEGFK FSKPIGDFKF GVSSESKPEE VKKDSKNDNF KFGLSSGLSN PVUA |
-Experimental information
| Beam | Instrument name: PETRA III EMBL P12 / City: Hamburg / 国: Germany / Type of source: X-ray synchrotron / Wavelength: 0.1 Å / Dist. spec. to detc.: 3 mm | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Detector | Name: Pilatus 2M | |||||||||||||||||||||||||||||||||
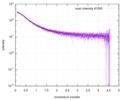
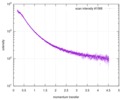
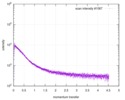
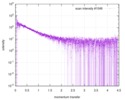
| Scan |  Measurement date: Nov 8, 2013 / Cell temperature: 23 °C / Exposure time: 0.05 sec. / Number of frames: 20 / Unit: 1/nm /
| |||||||||||||||||||||||||||||||||
| Distance distribution function P(R) | 
| |||||||||||||||||||||||||||||||||
| Result |  Comments: The protein contains a penultimate non-canonical amino acid p-acetylphenylalanine (207 Da) that is represented as U (selenocysteine, 168 Da) in the amino acid sequence for the entry. ...Comments: The protein contains a penultimate non-canonical amino acid p-acetylphenylalanine (207 Da) that is represented as U (selenocysteine, 168 Da) in the amino acid sequence for the entry. Therefore, the calculated MW from sequence (MW(expected)) must be adjusted accordingly (ca. 40 Da).
|
