 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | 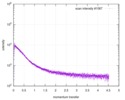 |
|---|---|
 Sample Sample | Unlabeled nucleoporin NSP1 (NSP) without denaturant
|
| Function / homology |  Function and homology information Function and homology informationnuclear pore central transport channel / Regulation of Glucokinase by Glucokinase Regulatory Protein / : / Regulation of HSF1-mediated heat shock response / nuclear pore nuclear basket / tRNA export from nucleus / SUMOylation of SUMOylation proteins / structural constituent of nuclear pore / SUMOylation of RNA binding proteins / RNA export from nucleus ...nuclear pore central transport channel / Regulation of Glucokinase by Glucokinase Regulatory Protein / : / Regulation of HSF1-mediated heat shock response / nuclear pore nuclear basket / tRNA export from nucleus / SUMOylation of SUMOylation proteins / structural constituent of nuclear pore / SUMOylation of RNA binding proteins / RNA export from nucleus / SUMOylation of chromatin organization proteins / nucleocytoplasmic transport / NLS-bearing protein import into nucleus / poly(A)+ mRNA export from nucleus / ribosomal large subunit export from nucleus / nuclear pore / ribosomal small subunit export from nucleus / nuclear periphery / phospholipid binding / protein import into nucleus / nuclear envelope / nuclear membrane Similarity search - Function |
| Biological species |  |
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2017 Title: Decoupling of size and shape fluctuations in heteropolymeric sequences reconciles discrepancies in SAXS vs. FRET measurements. Authors: Gustavo Fuertes / Niccolò Banterle / Kiersten M Ruff / Aritra Chowdhury / Davide Mercadante / Christine Koehler / Michael Kachala / Gemma Estrada Girona / Sigrid Milles / Ankur Mishra / ...Authors: Gustavo Fuertes / Niccolò Banterle / Kiersten M Ruff / Aritra Chowdhury / Davide Mercadante / Christine Koehler / Michael Kachala / Gemma Estrada Girona / Sigrid Milles / Ankur Mishra / Patrick R Onck / Frauke Gräter / Santiago Esteban-Martín / Rohit V Pappu / Dmitri I Svergun / Edward A Lemke /     Abstract: Unfolded states of proteins and native states of intrinsically disordered proteins (IDPs) populate heterogeneous conformational ensembles in solution. The average sizes of these heterogeneous ...Unfolded states of proteins and native states of intrinsically disordered proteins (IDPs) populate heterogeneous conformational ensembles in solution. The average sizes of these heterogeneous systems, quantified by the radius of gyration ( ), can be measured by small-angle X-ray scattering (SAXS). Another parameter, the mean dye-to-dye distance ( ) for proteins with fluorescently labeled termini, can be estimated using single-molecule Förster resonance energy transfer (smFRET). A number of studies have reported inconsistencies in inferences drawn from the two sets of measurements for the dimensions of unfolded proteins and IDPs in the absence of chemical denaturants. These differences are typically attributed to the influence of fluorescent labels used in smFRET and to the impact of high concentrations and averaging features of SAXS. By measuring the dimensions of a collection of labeled and unlabeled polypeptides using smFRET and SAXS, we directly assessed the contributions of dyes to the experimental values and For chemically denatured proteins we obtain mutual consistency in our inferences based on and , whereas for IDPs under native conditions, we find substantial deviations. Using computations, we show that discrepant inferences are neither due to methodological shortcomings of specific measurements nor due to artifacts of dyes. Instead, our analysis suggests that chemical heterogeneity in heteropolymeric systems leads to a decoupling between and that is amplified in the absence of denaturants. Therefore, joint assessments of and combined with measurements of polymer shapes should provide a consistent and complete picture of the underlying ensembles. |
 Contact author Contact author |
|
- Structure visualization
Structure visualization
- Downloads & links
Downloads & links
-Data source
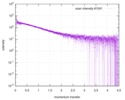
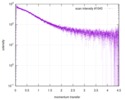
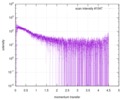
| SASBDB page |  SASDEC3 SASDEC3 |
|---|
-Related structure data
| Related structure data | C: citing same article ( |
|---|---|
| Similar structure data |
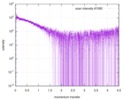
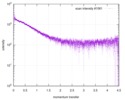
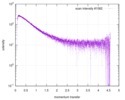
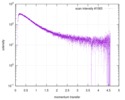





-External links
| Related items in Molecule of the Month |
|---|


-Models
-Sample
| Sample | Name: Unlabeled nucleoporin NSP1 (NSP) without denaturant / Specimen concentration: 2.00-5.00 |
|---|---|
| Buffer | Name: PBS, 10 mM DTT / pH: 7.4 |
| Entity #1249 | Type: protein / Description: Nucleoporin NSP1 / Formula weight: 17.635 / Num. of mol.: 1 Source: Saccharomyces cerevisiae (strain ATCC 204508 / S288c) References: UniProt: P14907 Sequence: GCNFNTPQQN KTPFSFGTAN NNSNTTNQNS STGAGAFGTG QSTFGFNNSA PNNTNNANSS ITPAFGSNNT GNTAFGNSNP TSNVFGSNNS TTNTFGSNSA GTSLFGSSSA QQTKSNGTAG GNTFGSSSLF NNSTNSNTTK PAFGGLNFGG GNNTTPSSTG NANTSNNLFG ATANANUA |
-Experimental information
| Beam | Instrument name: PETRA III EMBL P12 / City: Hamburg / 国: Germany / Type of source: X-ray synchrotron / Wavelength: 0.1 Å / Dist. spec. to detc.: 3 mm | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Detector | Name: Pilatus 2M | |||||||||||||||||||||||||||||||||
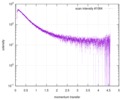
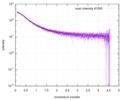
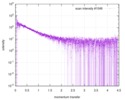
| Scan | 
| |||||||||||||||||||||||||||||||||
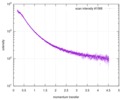
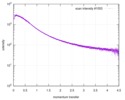
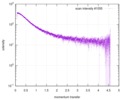
| Distance distribution function P(R) | 
| |||||||||||||||||||||||||||||||||
| Result |  Comments: The protein contains a penultimate non-canonical amino acid p-acetylphenylalanine (207 Da) that is represented as U (selenocysteine, 168 Da) in the amino acid sequence for the entry. ...Comments: The protein contains a penultimate non-canonical amino acid p-acetylphenylalanine (207 Da) that is represented as U (selenocysteine, 168 Da) in the amino acid sequence for the entry. Therefore, the calculated MW from sequence (MW(expected)) must be adjusted accordingly (ca. 40 Da).
|
